Nuove pubblicazioni
Nuove scoperte contribuiscono a una migliore comprensione delle cause della sindrome di Rett
Ultima recensione: 02.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.

La sindrome di Rett è un raro disturbo dello sviluppo neurologico per il quale attualmente non esiste una cura o un trattamento efficace. Causa gravi sintomi fisici e cognitivi, molti dei quali si sovrappongono ai disturbi dello spettro autistico.
La sindrome di Rett è causata da mutazioni nel gene MECP2, che è altamente espresso nel cervello e sembra svolgere un ruolo importante nel mantenimento della salute dei neuroni. Il gene è localizzato sul cromosoma X e la sindrome colpisce principalmente le ragazze. Per sviluppare trattamenti per la sindrome di Rett, i ricercatori vogliono comprendere meglio MECP2 e le sue funzioni nel cervello.
I ricercatori, tra cui il co-fondatore del Whitehead Institute Rudolf Jaenisch, studiano MECP2 da decenni, eppure molti aspetti fondamentali del gene rimangono sconosciuti. La proteina codificata dal gene, MECP2, è coinvolta nella regolazione genica; si lega al DNA e influenza i livelli di espressione di vari altri geni, ovvero la quantità di proteine che producono.
Tuttavia, i ricercatori non avevano un elenco completo dei geni interessati da MECP2 e non c'era consenso sul modo in cui MECP2 influisce su questi geni.
I primi studi su MECP2 suggerivano che agisse come un repressore, riducendo l'espressione dei suoi geni bersaglio, ma le ricerche di Jaenisch e altri avevano precedentemente dimostrato che MECP2 agisce anche come attivatore, aumentando l'espressione dei suoi geni bersaglio – e che potrebbe essere un attivatore in primo luogo. Era inoltre sconosciuto il meccanismo d'azione di MECP2, ovvero cosa esattamente la proteina faccia per causare cambiamenti nell'espressione genica.
I limiti della tecnologia hanno impedito ai ricercatori di fare chiarezza su questi interrogativi. Ma Yanish, il postdoc del suo laboratorio Yi Liu e l'ex membro del laboratorio di Yanish Anthony Flamier, ora professore associato presso il centro di ricerca CHU Sainte-Justine dell'Université de Montréal, hanno utilizzato tecniche all'avanguardia per rispondere a questi interrogativi rimanenti su MECP2 e acquisire nuove conoscenze sul suo ruolo nella salute e nelle patologie cerebrali.
I loro risultati sono stati pubblicati sulla rivista Neuron e i ricercatori hanno anche creato un archivio online dei loro dati MECP2, il portale MECP2-NeuroAtlas, come risorsa per altri ricercatori.
"Credo che questo studio cambierà radicalmente la comprensione di come MECP2 causi la sindrome di Rett. Abbiamo una comprensione completamente nuova del meccanismo e potrebbe aprire nuove strade per lo sviluppo di trattamenti per la malattia", afferma Janisch, che è anche professore di biologia al MIT.
Una comprensione più approfondita di MECP2 nel cervello
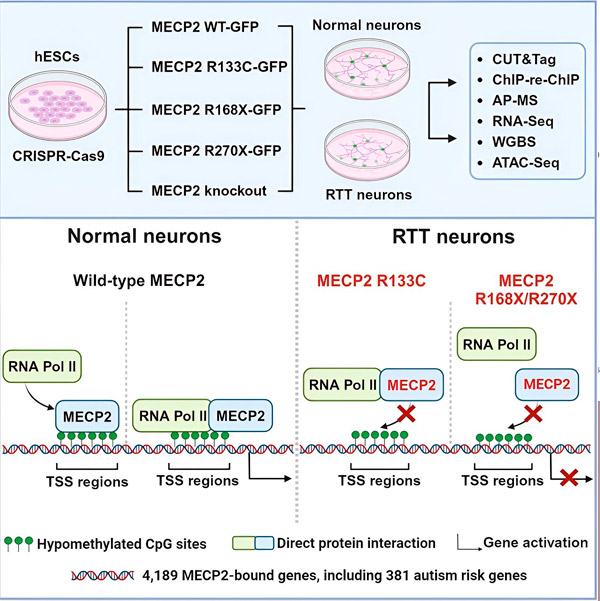
I ricercatori hanno innanzitutto creato una mappa dettagliata dei punti in cui MECP2 si lega nelle sequenze geniche neuronali umane, sia all'interno dei geni che nelle regioni regolatrici del DNA adiacenti. Hanno utilizzato un approccio chiamato CUT&Tag, in grado di individuare con elevata precisione le interazioni proteiche con il DNA.
I ricercatori hanno trovato più di 4.000 geni associati a MECP2. Hanno ripetuto la mappatura nei neuroni con mutazioni comuni di MECP2 associate alla sindrome di Rett per determinare dove MECP2 sia depleto durante la malattia.
Conoscere i geni a cui si lega MECP2 ha permesso a Liu e Flamier di iniziare a stabilire connessioni tra i bersagli di MECP2 e la salute del cervello. Hanno scoperto che molti dei suoi bersagli sono coinvolti nello sviluppo e nella funzione degli assoni e delle sinapsi neuronali.
Hanno inoltre confrontato il loro elenco di target MECP2 con il database dei geni associati all'autismo della Simons Foundation Autism Research Initiative (SFARI) e hanno scoperto che 381 geni in quel database sono target MECP2.
Fonte: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
Questi risultati potrebbero aiutare a chiarire i meccanismi alla base dei sintomi autistici nella sindrome di Rett e fornire un buon punto di partenza per indagare il possibile ruolo di MECP2 nell'autismo.
"Abbiamo creato la prima mappa integrata dell'epigenoma di MECP2 in condizioni di salute e malattia, e questa mappa può orientare la ricerca futura", afferma Liu. "Sapere quali geni sono bersaglio di MECP2 e quali geni sono direttamente compromessi nella malattia fornisce una solida base per comprendere la sindrome di Rett e porre domande sulla regolazione genica nei neuroni."
I ricercatori hanno anche esaminato se MECP2 aumentasse o diminuisse l'espressione dei suoi geni bersaglio. Coerentemente con la storia di MECP2, identificata da alcuni come un attivatore e da altri come un repressore, Liu e Flamier hanno trovato esempi in cui MECP2 svolgeva entrambi i ruoli.
Tuttavia, mentre MECP2 è più spesso considerato un repressore, Liu e Flamier hanno scoperto che è principalmente un attivatore, confermando i risultati precedenti di Jaenisch e Liu. Un nuovo esperimento ha dimostrato che MECP2 attiva almeno l'80% dei suoi bersagli, e un altro ha scoperto che attiva fino all'88%.
La mappa dei geni bersaglio creata dai ricercatori ha fornito ulteriori informazioni sul ruolo di MECP2 come attivatore. Hanno scoperto che, per i geni attivati da MECP2, questo si lega tipicamente a una regione del DNA a monte del gene, chiamata sito di inizio della trascrizione.
Questo è il sito in cui i meccanismi cellulari avviano il processo di trascrizione di un gene in RNA, dopodiché l'RNA viene tradotto in una proteina funzionale, che è il prodotto dell'espressione genica. La presenza di MECP2 nel sito di inizio della trascrizione, dove inizia l'espressione genica, è coerente con il suo ruolo di attivatore genico.
I ricercatori si sono quindi prefissati di determinare il ruolo di MECP2 nell'attivazione genica. Hanno esaminato a quali molecole MECP2 si lega in questo sito, oltre al DNA, e hanno scoperto che MECP2 interagisce direttamente con un complesso proteico chiamato RNA polimerasi II (RNA Pol II). L'RNA Pol II è una macchina cellulare chiave che trascrive il DNA in RNA. L'RNA Pol II non è in grado di trovare geni da sola, quindi necessita di una varietà di cofattori, o collaboratori proteici, per svolgere il suo compito.
I ricercatori ipotizzano che MECP2 agisca come uno di questi cofattori, aiutando l'RNA Pol II a iniziare la trascrizione nei geni in cui MECP2 si lega. L'analisi strutturale di MECP2 ha identificato parti della molecola che si legano all'RNA Pol II, e altri esperimenti hanno confermato che la perdita di MECP2 riduce la presenza di RNA Pol II nei siti di inizio della trascrizione appropriati, nonché i livelli di espressione dei geni bersaglio.
Ciò suggerisce che la sindrome di Rett possa essere causata da una ridotta trascrizione dei geni bersaglio di MECP2, dovuta a mutazioni di MECP2 che ne impediscono il legame con la RNA Pol II o con il DNA. In linea con questa idea, le mutazioni di MECP2 più comuni associate alla malattia sono troncamenti: mutazioni in cui manca una parte della proteina, che possono alterare l'interazione tra MECP2 e RNA Pol II.
I ricercatori sperano che le loro scoperte non solo cambieranno la nostra comprensione di MECP2, ma che una comprensione più approfondita e ampia di come MECP2 influenza lo sviluppo e la funzione del cervello potrebbe portare a nuove intuizioni che aiuteranno le persone con sindrome di Rett e disturbi correlati, tra cui l'autismo.
"Questo progetto è un ottimo esempio della natura collaborativa del laboratorio Janisch", afferma Flamier. "Rudolf ed io avevamo un problema specifico legato alla sindrome di Rett, e io avevo esperienza con la tecnologia CUT&Tag, che poteva risolvere il problema. Attraverso il confronto, ci siamo resi conto che potevamo unire i nostri sforzi e ora disponiamo di un'ampia raccolta di informazioni su MECP2 e sui suoi legami con le malattie."