Esperto medico dell'articolo

Nuove pubblicazioni
Malattia del tessuto connettivo misto: cause, sintomi, diagnosi, trattamento
Ultima recensione: 07.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
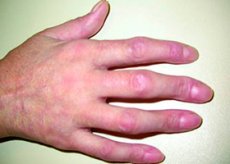
La connettivite mista è una malattia rara caratterizzata dalla presenza simultanea di manifestazioni di lupus eritematoso sistemico, sclerosi sistemica, polimiosite o dermatomiosite e artrite reumatoide con titoli molto elevati di autoanticorpi antinucleo circolanti contro le ribonucleoproteine (RNP). Sono caratteristici lo sviluppo di edema alle mani, fenomeno di Raynaud, poliartralgia, miopatia infiammatoria, ipotensione esofagea e compromissione della funzionalità polmonare. La diagnosi si basa sull'analisi del quadro clinico della malattia e sulla rilevazione di anticorpi contro le RNP in assenza di anticorpi caratteristici di altre malattie autoimmuni. Il trattamento è simile a quello del lupus eritematoso sistemico e prevede l'uso di glucocorticoidi per la malattia da moderata a grave.
La malattia mista del tessuto connettivo (MCTD) è presente in tutto il mondo e in tutte le etnie, con un picco di incidenza nell'adolescenza e nella seconda decade di vita.
 [
[ Manifestazioni cliniche della malattia mista del tessuto connettivo
Il fenomeno di Raynaud può precedere di diversi anni altre manifestazioni della malattia. Spesso, le prime manifestazioni della connettivite mista possono assomigliare all'esordio di lupus eritematoso sistemico, sclerodermia, artrite reumatoide, polimiosite o dermatomiosite. Tuttavia, indipendentemente dalla natura delle manifestazioni iniziali della malattia, questa tende a progredire e diffondersi con un cambiamento nella natura delle manifestazioni cliniche.
L'edema più comune interessa le mani, soprattutto le dita, conferendole un aspetto a salsiccia. Le alterazioni cutanee sono simili a quelle osservate nel lupus o nella dermatomiosite. Lesioni simili a quelle osservate nella dermatomiosite, così come necrosi ischemica e ulcerazione della punta delle dita, sono meno comuni.
Quasi tutti i pazienti lamentano poliartralgia, il 75% presenta segni evidenti di artrite. L'artrite di solito non porta a cambiamenti anatomici, ma possono verificarsi erosioni e deformazioni, come nell'artrite reumatoide. Si osserva spesso debolezza dei muscoli prossimali, sia con che senza dolore.
Il danno renale si verifica in circa il 10% dei pazienti ed è spesso lieve, ma in alcuni casi può portare a complicazioni e al decesso. Nella connettivite mista, la neuropatia sensoriale del nervo trigemino si sviluppa più frequentemente rispetto ad altre connettiviti.
Diagnosi della malattia mista del tessuto connettivo
La connettivite mista deve essere sospettata in tutti i pazienti con LES, sclerodermia, polimiosite o AR, qualora si sviluppino ulteriori manifestazioni cliniche. Innanzitutto, è necessario testare la presenza di anticorpi antinucleo (ARA), anticorpi contro l'antigene nucleare estraibile e RNP. Se i risultati ottenuti sono compatibili con una possibile MCDT (ad esempio, titolo molto elevato di anticorpi contro l'RNA), è necessario testare le gammaglobuline, il complemento, il fattore reumatoide, gli anticorpi contro l'antigene Jo-1 (istidil-tRNA sintetasi), gli anticorpi contro la componente resistente alla ribonucleasi dell'antigene nucleare estraibile (Sm) e la doppia elica del DNA per escludere altre patologie. Il piano di ulteriori indagini dipende dalla presenza di sintomi di coinvolgimento di organi e sistemi: miosite, coinvolgimento renale e polmonare richiedono l'implementazione di metodi diagnostici appropriati (in particolare, risonanza magnetica, elettromiografia, biopsia muscolare).
Quasi tutti i pazienti presentano titoli elevati (spesso >1:1000) di anticorpi antinucleari rilevati mediante fluorescenza. Gli anticorpi contro l'antigene nucleare estraibile sono solitamente presenti a titoli molto elevati (>1:100.000). Gli anticorpi contro l'RNP sono tipicamente presenti, mentre gli anticorpi contro la componente Sm dell'antigene nucleare estraibile sono assenti.
Il fattore reumatoide può essere rilevato a titoli piuttosto elevati. La VES è spesso elevata.
Prognosi e trattamento della malattia mista del tessuto connettivo
La sopravvivenza a dieci anni è dell'80%, ma la prognosi dipende dalla gravità dei sintomi. Le principali cause di morte sono ipertensione polmonare, insufficienza renale, infarto del miocardio, perforazione del colon, infezioni disseminate ed emorragia cerebrale. Alcuni pazienti possono mantenere una remissione a lungo termine senza alcun trattamento.
Il trattamento iniziale e di mantenimento della connettivite mista è simile a quello del lupus eritematoso sistemico. La maggior parte dei pazienti con malattia da moderata a grave risponde alla terapia con glucocorticoidi, soprattutto se iniziata precocemente. La malattia lieve è controllata con successo con salicilati, altri FANS, antimalarici e, in alcuni casi, glucocorticoidi a basso dosaggio. Il grave coinvolgimento di organi e sistemi richiede glucocorticoidi ad alto dosaggio (ad es., prednisolone 1 mg/kg una volta al giorno, per via orale) o immunosoppressori. In caso di sviluppo di sclerosi sistemica, viene somministrato un trattamento appropriato.