Esperto medico dell'articolo

Nuove pubblicazioni
Sindrome di Usher
Ultima recensione: 04.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
La sindrome di Usher è una malattia ereditaria che si manifesta con sordità completa fin dalla nascita e cecità progressiva con l'età. La perdita della vista è associata alla retinite pigmentosa, un processo di degenerazione pigmentaria della retina. Molte persone con sindrome di Usher presentano anche gravi problemi di equilibrio.
Epidemiologia
Grazie alla ricerca, è stato possibile stabilire che la sindrome di Usher colpisce circa l'8% dei bambini sordomuti esaminati (i test sono stati condotti in istituti specializzati per sordomuti). La retinite pigmentosa è stata osservata nel 6-10% dei pazienti affetti da sordità congenita, che a sua volta si riscontra in circa il 30% delle persone con retinopatia pigmentosa.
Si ritiene che questa malattia si manifesti in circa 3-10 persone su 100.000 in tutto il mondo. Può essere osservata in egual misura sia nelle donne che negli uomini. Circa il 5-6% della popolazione mondiale soffre di questa sindrome. Circa il 10% di tutti i casi di sordità profonda infantile si verifica a causa della sindrome di Usher di tipo I e II.
Negli Stati Uniti, i tipi 1 e 2 sono i più comuni. Insieme, rappresentano circa il 90-95% di tutti i casi di sindrome di Usher nei bambini.
Le cause Sindrome di Usher
La sindrome di Usher di tipo I, II e III ha una causa autosomica recessiva, mentre il tipo IV è considerato un disturbo del cromosoma X. Le cause di cecità e sordità associate a questa sindrome non sono ancora state sufficientemente studiate. Si presume che le persone affette da questa malattia siano ipersensibili a componenti che possono danneggiare la struttura del DNA. Inoltre, questa malattia può essere associata a disturbi del sistema immunitario, ma in questo caso non esiste un quadro preciso di questo processo.
Nel 1989, anomalie cromosomiche sono state identificate per la prima volta in pazienti con sindrome di tipo II, il che potrebbe in futuro portare a un metodo per isolare i geni che causano la sindrome. Potrebbe anche essere possibile identificare questi geni nei portatori e sviluppare specifici test genetici prenatali.
 [ 8 ]
[ 8 ]
Fattori di rischio
La sindrome è ereditaria quando entrambi i genitori sono affetti, ovvero è ereditata con un gene recessivo. Un bambino può ereditare la malattia anche se i suoi genitori sono portatori sani. Se entrambi i futuri genitori hanno questo gene, la probabilità di avere un figlio con questa sindrome è di 1 su 4. Una persona che ha un solo gene per la sindrome è considerata portatrice, ma non presenta sintomi del disturbo. Al giorno d'oggi, non è ancora possibile determinare se una persona ha il gene per questa malattia.
Se un bambino nasce da genitori uno dei quali non possiede tale gene, la probabilità che erediti la sindrome è molto bassa, ma sarà sicuramente portatore sano.
Sintomi Sindrome di Usher
I sintomi della sindrome di Usher includono perdita dell'udito e un accumulo anomalo di cellule pigmentate nelle strutture oculari. Il paziente sviluppa quindi una degenerazione della retina, che causa un deterioramento della vista e, nei casi più gravi, la perdita della vista.
La perdita dell'udito neurosensoriale può essere lieve o completa e di solito non progredisce dalla nascita. Tuttavia, la pigmentazione della retina può iniziare a svilupparsi durante l'infanzia o più tardi. I risultati dei test hanno dimostrato che l'acuità visiva centrale può essere mantenuta per molti anni, anche quando la visione periferica peggiora (una condizione chiamata "visione a tunnel").
Queste sono le principali manifestazioni della malattia, a cui talvolta possono aggiungersi altri disturbi, come psicosi e altri disturbi mentali, problemi all'orecchio interno e/o cataratta.
Forme
Nel corso della ricerca sono stati identificati 3 tipi di questa malattia, nonché una quarta forma piuttosto rara.
Il tipo I della malattia è caratterizzato da sordità completa congenita e disturbi dell'equilibrio. Spesso, questi bambini iniziano a camminare solo all'età di 1,5 anni. Il deterioramento della vista inizia solitamente all'età di 10 anni e lo sviluppo finale della cecità notturna inizia all'età di 20 anni. I bambini affetti da questo tipo di malattia possono sviluppare un progressivo deterioramento della visione periferica.
Nella patologia di tipo II si osserva una sordità moderata o congenita. In questo caso, il peggioramento della sordità parziale spesso non si verifica più. La retinite pigmentosa inizia a svilupparsi verso la fine dell'adolescenza o dopo i 20 anni. Lo sviluppo della cecità notturna inizia solitamente tra i 29 e i 31 anni. Il deficit visivo nella patologia di tipo II progredisce generalmente un po' più lentamente rispetto al tipo I.
Il tipo III della malattia è caratterizzato da una progressiva perdita dell'udito, che di solito inizia durante la pubertà, nonché dallo sviluppo graduale durante lo stesso periodo (leggermente più tardi della perdita dell'udito) di retinite pigmentosa, che può diventare un fattore nello sviluppo della cecità progressiva.
Le manifestazioni della patologia di tipo IV si verificano principalmente nei maschi. In questo caso, si osservano anche disturbi progressivi e perdita dell'udito e della vista. Questa forma è molto rara e di solito ha una natura cromosomica X.
Diagnostica Sindrome di Usher
La diagnosi della sindrome di Usher si basa sulla combinazione di sordità improvvisa e perdita progressiva della vista osservata nel paziente.
Test
Per rilevare la mutazione potrebbe essere richiesto uno speciale test genetico.
Sono stati individuati undici loci genetici che possono causare lo sviluppo della sindrome di Usher e sono stati identificati nove geni che sono sicuramente la causa del disturbo:
- Tipo 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Tipo 2: ush2a, VLGR1, WHRN.
- Sindrome di Usher tipo 3: USH3A.
Gli scienziati del NIDCD, insieme ai colleghi delle università di New York e Israele, hanno identificato una mutazione chiamata R245X nel gene Pcdh15, responsabile di un'elevata percentuale di sindrome di Usher di tipo 1 nella popolazione ebraica.
Per informazioni sui laboratori che eseguono sperimentazioni cliniche, visita https://www.genetests.org e cerca "sindrome di Usher" nell'elenco dei laboratori.
Per informazioni sugli studi clinici esistenti che includono test genetici per la sindrome di Usher, visitare https://www.clinicaltrials.gov e cercare "sindrome di Usher" o "test genetici per la sindrome di Usher".
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Diagnostica strumentale
Esistono diversi metodi di diagnosi strumentale:
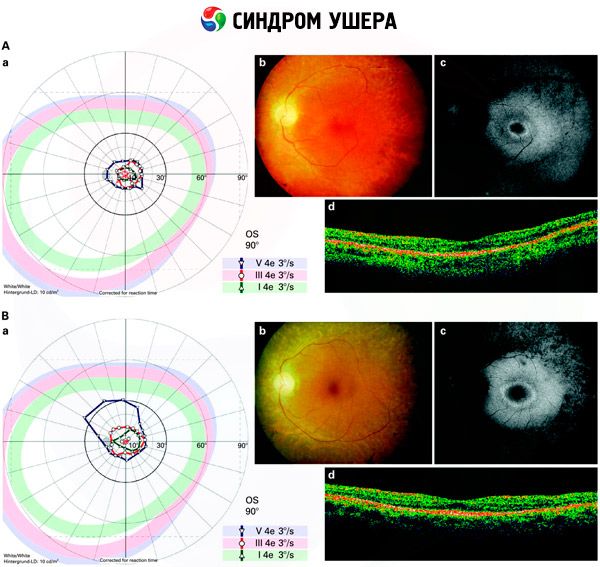
- Esame del fondo dell'occhio per rilevare la presenza di macchie pigmentate sulla retina, nonché il restringimento dei vasi retinici;
- Elettroretinogramma, che consente di rilevare le deviazioni degenerative iniziali della retina oculare. Mostra l'estinzione delle vie elettroradiografiche;
- L'elettronistagmogramma (ENG) misura i movimenti oculari involontari che potrebbero indicare la presenza di uno squilibrio.
- Audiometria, utilizzata per determinare la presenza di sordità e la sua gravità.
Diagnosi differenziale
La sindrome di Usher deve essere differenziata da alcuni disturbi simili.
Sindrome di Hallgren, caratterizzata da ipoacusia congenita e progressiva perdita della vista (si sviluppano anche cataratta e nistagmo). Ulteriori sintomi includono atassia, disturbi psicomotori, psicosi e ritardo mentale.
Sindrome di Alström, una malattia ereditaria in cui la retina degenera, con conseguente perdita della visione centrale. Questa sindrome è associata all'obesità infantile. Contemporaneamente, diabete mellito e perdita dell'udito iniziano a svilupparsi dopo i 10 anni.
La rosolia in una donna incinta nel primo trimestre può causare diverse anomalie nello sviluppo del bambino. Tra le conseguenze di tali anomalie figurano la perdita dell'udito, nonché (o) problemi alla vista, oltre a vari difetti dello sviluppo.
Chi contattare?
Trattamento Sindrome di Usher
Attualmente non esiste una cura per la sindrome di Usher. Pertanto, la terapia in questo caso consiste principalmente nel rallentare il processo di perdita della vista e nel compensare la perdita dell'udito. I possibili metodi di trattamento includono:
- Assunzione di vitamina A (alcuni oculisti ritengono che dosi elevate di palmitato di vitamina A possano rallentare, ma non arrestare, la progressione della retinite pigmentosa);
- Impianto di speciali dispositivi elettronici nelle orecchie del paziente (apparecchi acustici, impianti cocleari).
Gli oculisti raccomandano che la maggior parte degli adulti affetti da forme comuni di retinite pigmentosa assuma 15.000 UI (unità internazionali) di palmitato di vitamina A al giorno sotto supervisione. Poiché le persone con sindrome di Usher di tipo 1 non sono state incluse nello studio, dosi elevate di vitamina A non sono raccomandate per questo gruppo di pazienti. Chi sta prendendo in considerazione l'assunzione di vitamina A dovrebbe discutere questa opzione terapeutica con il proprio medico. Altre raccomandazioni per questa opzione terapeutica includono:
- Modificare la propria dieta includendo alimenti ricchi di vitamina A.
- Le donne che stanno pianificando una gravidanza dovrebbero interrompere l'assunzione di dosi elevate di vitamina A tre mesi prima del concepimento, a causa di un aumento del rischio di difetti alla nascita.
- Le donne incinte dovrebbero interrompere l'assunzione di dosi elevate di vitamina A a causa di un aumento del rischio di malformazioni congenite.
È inoltre importante adattare un bambino di questo tipo alla vita sociale. Ciò richiede l'aiuto di insegnanti di sostegno e psicologi. Nel caso in cui il paziente abbia iniziato ad avere una progressiva perdita della vista, è necessario insegnargli a usare il linguaggio dei segni.
Previsione
La sindrome di Usher ha una prognosi sfavorevole. Il campo visivo e la sua acuità visiva iniziano a deteriorarsi nell'arco di 20-30 anni nella maggior parte dei pazienti affetti da questa malattia, di qualsiasi tipo. In alcuni casi, si verifica una perdita completa della vista bilaterale. La perdita dell'udito, che è sempre accompagnata da mutismo, si evolve molto rapidamente in una perdita completa dell'udito bilaterale.