Esperto medico dell'articolo

Nuove pubblicazioni
Epilessia criptogenetica con convulsioni negli adulti
Ultima recensione: 23.04.2024

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
Secondo la classificazione internazionale valida fino allo scorso anno, i sintomi sintomatici o secondari, causati dalla sconfitta delle strutture cerebrali, idiopatica, primaria (malattia presumibilmente ereditaria ereditaria) e epilessia criptogenica sono stati isolati. Quest'ultima opzione significa che la diagnostica moderna non ha stabilito alcun motivo per le crisi epilettiche periodiche e anche la predisposizione ereditaria non viene tracciata. Il concetto stesso di "criptogenetico" è tradotto dal greco come "origine sconosciuta" (kryptos - segreto, segreto, genere nato).
La scienza non si ferma e, probabilmente, verrà stabilita l'origine di periodici attacchi epilettici di eziologia sconosciuta. Gli esperti suggeriscono che l'epilessia criptogenica è una malattia sintomatica secondaria, la cui genesi non può essere stabilita al livello attuale di diagnosi.
 [
[Epidemiologia
L'epilessia e le sindromi epilettiche sono patologie neurologiche molto comuni, oltre a causare spesso gravi conseguenze. La manifestazione di convulsioni epilettiche può verificarsi in persone di qualsiasi genere e a qualsiasi età. Si stima che circa il 5% della popolazione mondiale abbia avuto almeno un attacco nelle loro vite.
Ogni anno, la diagnosi di epilessia o sindrome epilettica viene mediamente valutata ogni 30-50 abitanti su 100mila persone che vivono sulla Terra. Il più delle volte le crisi epilettiche si verificano nei bambini (da 100 a 233 casi per 100 mila abitanti). Il picco della manifestazione cade nel periodo perinatale, quindi il tasso di incidenza è quasi dimezzato. I tassi più bassi in individui da 25 a 55 anni - circa 20-30 casi per 100 mila della popolazione. Quindi aumenta la probabilità di attacchi epilettici e dall'età di 70 anni l'incidenza varia da 150 casi o più per 100.000 persone.
Le cause dell'epilessia sono stabilite in circa il 40% dei casi, quindi la malattia di eziologia sconosciuta non è rara. Gli spasmi infantili (sindrome di West), correlati all'epilessia criptogenetica, sono diagnosticati in bambini di età compresa tra 4 e 6 mesi, un bambino con una diagnosi tale che si verifica in media tra 3200 neonati.
Le cause epilessia crittogena
La base per la diagnosi di epilessia sono crisi ricorrenti causate da diventare anormalmente forte scarica elettrica risultante dall'attività sincronizzazione delle cellule cerebrali in tutte le bande di frequenza che esteriormente manifestano nella comparsa sensomotorio, neurologiche e sintomi mentali.
Per l'emergenza di un attacco epilettico, è necessaria la presenza di cosiddetti neuroni epilettici, caratterizzati dall'instabilità del potenziale di riposo (la differenza di potenziale della cellula non eccitata sui lati interno ed esterno della membrana). Di conseguenza, il potenziale d'azione di un neurone epilettico eccitato ha un'ampiezza, una durata e una frequenza sostanzialmente più alte del normale, il che porta allo sviluppo di un attacco epilettico. Si ritiene che le convulsioni si verificano in persone che hanno una predisposizione ereditaria a tali cambiamenti, cioè gruppi di neuroni epilettici che sono in grado di sincronizzare la loro attività. I focolai epilettici si formano anche in luoghi del cervello con struttura alterata a causa di lesioni, infezioni, intossicazioni, sviluppo di tumori.
Quindi, nei pazienti a cui viene diagnosticata l'epilessia criptogenetica, i moderni metodi di neuroimaging non rivelano alcun disturbo nella struttura della sostanza cerebrale e non ci sono epilettici nella storia familiare. Tuttavia, i pazienti spesso hanno crisi epilettiche abbastanza frequenti di vario tipo, che sono difficili da trattare (forse, solo perché la loro causa non è chiara).
Di conseguenza, i noti fattori di rischio per l'insorgenza di crisi epilettiche - genetica, disordine della struttura cerebrale, processi metabolici nei suoi tessuti, conseguenze di ferite alla testa o processi infettivi durante indagini e sondaggi non vengono rilevati.
Secondo la nuova classificazione dell'epilessia nel 2017, si distinguono sei categorie eziologiche della malattia. Anziché sintomatico, si consiglia ora di determinare il tipo di epilessia per una ragione consolidata: strutturale, infettiva, metabolica, immunitaria o una combinazione di essi. L'epilessia idiopatica presupponeva la presenza di una predisposizione ereditaria ed è ora chiamata genetica. Il termine "criptogenetico" è sostituito da "un fattore etiologico sconosciuto", perché il significato della dicitura è diventato più comprensibile, ma non è cambiato.
La patogenesi dell'epilessia presumibilmente simile a questo: la formazione del focus epilettico, cioè comunità di neuroni con compromissione della creazione elettrogenesi → nel cervello dei sistemi epilettiche (con eccesso di rilascio dei neurotrasmettitori eccitatori lanciato "glutammato a cascata', colpisce tutti i nuovi neuroni e promuove la formazione di nuovi focolai epileptogeneza) → formazione di patologica connessioni interneuronali → si verifica generalizzazione dell'epilessia.
L'ipotesi principale del meccanismo di sviluppo epilessia si presume che il processo patologico inizia un disturbo dello stato di equilibrio tra neurotrasmettitori eccitatori (glutammato, aspartato) e responsabili dei processi di inibizione (γ-aminobutirrico, taurina, glicina, noradrenalina, dopamina, serotonina). Ciò che viola esattamente questo equilibrio nel nostro caso rimane sconosciuto. Tuttavia, le membrane cellulari affetti di neuroni, disturbato cinetica del flusso di ioni - inattivati pompe ioniche, dall'altro, canali ionici attivati disturbate concentrazione intracellulare di ioni positivi di potassio, sodio e cloro. Destrutturato patologico con membrana a scambio ionico determina le variazioni del livello del flusso sanguigno cerebrale. Disfunzione del recettore del glutammato e la produzione di autoanticorpi che provoca convulsioni. Ricorrente Neural livello eccessivamente intenso realizzato in forma di convulsioni, che portano a profonde alterazioni dei processi metabolici nelle cellule del cervello e sostanza provocano lo sviluppo del successivo sequestro.
La specificità di questo processo è l'aggressività dei neuroni del focus epilettico in relazione alle aree invariate del cervello, che consente loro di subordinare nuovi siti. La creazione di sistemi epilettici avviene durante la formazione di interrelazioni patologiche tra il focus epilettico e le componenti strutturali del cervello, in grado di attivare il meccanismo di sviluppo dell'epilessia. A tali strutture appartengono: il talamo, il sistema limbico, la formazione reticolare della parte centrale del tronco cerebrale. Le interrelazioni che insorgono con il cervelletto, il nucleo caudato della subcorteccia, la corteccia orbitale anteriore, al contrario, rallentano lo sviluppo dell'epilessia.
Nel corso dello sviluppo della malattia, si forma un sistema patologico chiuso: il cervello epilettico. La sua educazione è completata dal disturbo del metabolismo cellulare e dall'interazione dei neurotrasmettitori, dalla circolazione cerebrale, dalla crescita dell'atrofia e dei vasi cerebrali, dall'attivazione di specifici processi autoimmuni cerebrali.
Sintomi epilessia crittogena
La principale manifestazione clinica di questa malattia è il sequestro epilettico. Il sospetto di epilessia si verifica quando il paziente ha visto almeno due crisi epilettiche riflesse (non provocate), le cui manifestazioni sono molto diverse. Ad esempio, gli attacchi epileptopodobnye causati da alte temperature e non si verificano nello stato normale, l'epilessia non lo sono.
Nei pazienti con epilessia criptogenetica, possono verificarsi convulsioni di diverso tipo e abbastanza spesso.
I primi segni dello sviluppo della malattia (prima della comparsa di crisi epilettiche a pieno titolo) possono rimanere inosservati. Nel gruppo a rischio, le persone che hanno sofferto di convulsioni febbrili nella prima infanzia, con una conclusione sull'aumento della prontezza convulsiva. Nel periodo prodromico si possono osservare disturbi del sonno, irritabilità aumentata, labilità emotiva.
Inoltre, non sempre gli attacchi hanno luogo nella classica forma generalizzata con caduta, convulsioni, perdita di coscienza.
A volte i primi segni sono disturbi del linguaggio, un paziente in coscienza, ma non parla e non risponde alle domande, o sviene periodici di breve durata. Questo non dura a lungo - un paio di minuti, quindi è lasciato senza attenzione.
Più facilmente, si verificano semplici crisi focali o parziali (locali, limitate), le cui manifestazioni dipendono dalla posizione del focus epilettico. Il paziente non perde conoscenza al momento del parossismo.
Durante un semplice attacco motorio, si possono osservare tic, contrazioni agli arti, crampi muscolari, movimenti rotazionali del tronco e della testa. Il paziente può emettere suoni inarticolati o tacere, senza rispondere alle domande, schiaffeggiare, leccare, fare movimenti di masticazione.
Le semplici convulsioni sensoriali sono caratterizzate da parestesie - intorpidimento di varie parti del corpo, gusto insolito o sensazioni olfattive, solitamente spiacevoli; disturbi visivi - lampi di luce, mesh, mosche davanti agli occhi, visione a tunnel.
Parossismi vegetativi si verificano improvvisi pallore o iperemia della pelle, aumento della frequenza cardiaca, la pressione sanguigna, il battito cardiaco, la contrazione o l'espansione delle pupille, fastidio allo stomaco fino a quando il dolore e il vomito.
Le convulsioni mentali si manifestano con derealizzazione / depersonalizzazione, attacchi di panico. Di regola, sono precursori di complesse crisi focali, che sono già associate a una violazione della coscienza. Il paziente si rende conto di avere un attacco, ma non può chiedere aiuto. Eventi che si sono verificati durante l'attacco, cancellati dalla memoria del paziente. Le funzioni cognitive umane sono disturbate - c'è una sensazione di irrealtà di ciò che sta accadendo, nuovi cambiamenti dentro di sé.
Le convulsioni focali con successiva generalizzazione iniziano come semplici (complesse), trasformandosi in parossismi tonico-clonici generalizzati. Passano tre minuti e passano in un sonno profondo.
Le convulsioni generalizzate si verificano in una forma più grave e si dividono in:
- tonico-clonico, che si verifica in questa sequenza - il paziente perde coscienza, cade, il suo corpo si flette e allunga un arco, le contrazioni convulsive dei muscoli iniziano in tutto il corpo; gli occhi del paziente si alzano, le sue pupille sono dilatate in questo momento; il paziente urla, trasformando blu come conseguenza della cessazione della respirazione per pochi secondi, v'è una salivazione schiumoso (schiuma può acquisire un colore rosato per la presenza di sangue in esso, indicando che la lingua o la guancia mordere); A volte c'è uno svuotamento involontario della vescica;
- mioclonica appaiono come intermittente (ritmica e spasmodica) contrazione dei muscoli per alcuni secondi su tutto il corpo o parti del corpo individuale che assomigliano a gambe svolazzanti, squat, spremere le mani a pugno e altri movimenti ripetitivi; La coscienza, soprattutto con crisi focali, persiste (più spesso questa specie si osserva durante l'infanzia);
- assenze - convulsioni non convulsivo con un breve (5-20 secondi) spegnimento della coscienza, dovute al fatto che una persona muore con occhi inespressivi aperti, e non risponde agli stimoli normalmente non cade, tornando in sé, ha proseguito il lavoro interrotto, e non ricorda una vestibilità ;
- Le assenze atipiche sono accompagnate da cadute, svuotamento involontario della vescica, più prolungato e si verificano in forme gravi della malattia, in combinazione con ritardo mentale e altri sintomi di disturbi mentali;
- convulsioni atone (acinetici) - il paziente cade bruscamente a seguito della perdita del tono muscolare (con epilessia focale - può essere atonia singoli gruppi muscolari: viso - cedimenti della mandibola, il collo - il paziente è seduto o in piedi, con la testa appesa), la durata di un attacco meno di un minuto; L'atonia alle assenze inizia gradualmente - il paziente si assesta lentamente, con attacchi atonici isolati - cade bruscamente.
Nel periodo post-fatale il paziente è pigro e rallentato, se non per frenare, si addormenta (soprattutto dopo quelli generalizzati).
I tipi di epilessia corrispondono ai tipi di convulsioni. Le convulsioni (parziali) focali si sviluppano a livello epilettico locale, quando una scarica eccessivamente intensa incontra resistenza nelle aree vicine e si estingue, non diffondendosi ad altre parti del cervello. In questi casi, viene diagnosticata l'epilessia focale criptogenetica.
Il decorso clinico della malattia con un focus epilettico limitato (forma focale) è determinato dalla posizione della sua localizzazione.
La lesione più spesso osservata della regione temporale. La corrente di questa forma è progrediente, le crisi spesso hanno un tipo misto, continuano per diversi minuti. Epilessia temporale criptogenetica al di fuori degli attacchi si manifesta con mal di testa, vertigini costanti, nausea. I pazienti con questa forma di localizzazione si lamentano di minzione frequente. Prima di una crisi, i pazienti sentono l'aura-precursore.
La lesione può essere localizzata nel lobo frontale del cervello. Le convulsioni sono caratterizzate da repentinità senza un'aura prodromica. Il paziente ha la contrazione della sua testa, gli occhi gli rotolano sotto la fronte e, a parte, i suoi gesti automatici sono piuttosto complicati. Il paziente può perdere conoscenza, cade, ha spasmi muscolari tonico-clonici su tutto il corpo. Con questa localizzazione, ci sono una serie di sequestri di breve durata, a volte con una transizione generalizzata e / o epistatus. Possono iniziare non solo durante la veglia diurna, ma durante la notte dormono. L'epilessia frontale criptogenica, che si sviluppa, provoca disturbi mentali (pensiero violento, derealizzazione) e il sistema nervoso autonomo.
Sequestri di tipo touch (sensazione di movimento di aria calda nella pelle, tocco leggero) combinata con una strappi delle parti del corpo, la parola e disturbi motori atonia, accompagnato da incontinenza urinaria.
La localizzazione del focus epilettico nella regione orbitale frontale appare allucinazioni olfattive, ipersalivazione, fastidio epigastrico, così - disturbi del linguaggio, tosse e gonfiore della laringe.
Se le cascate di iperattività elettrica si diffondono a tutte le parti del cervello, si sviluppa una crisi generalizzata. In questo caso, al paziente viene diagnosticata un'epilessia criptogenica generalizzata. In questo caso, le convulsioni sono caratterizzate da intensità, perdita di coscienza e risultato nel sonno prolungato di un paziente. Al risveglio, i pazienti si lamentano di mal di testa, fenomeni visivi, debolezza e devastazione.
Ci sono anche combinati (quando si verificano sia crisi focali e generalizzate) che un tipo sconosciuto di epilessia.
L'epilessia criptogenetica negli adulti è considerata, e non senza motivi, secondaria a un fattore eziologico non identificato. Caratterizzato dall'improvvisa convulsioni. Al di fuori delle manifestazioni dei sintomi clinici, gli epilettici hanno una psiche instabile, un temperamento esplosivo, una tendenza all'aggressività. Di solito la malattia inizia con la manifestazione di alcune forme focali. Man mano che la malattia si sviluppa, i fuochi della lesione si diffondono ad altre parti del cervello, per uno stadio trascurato, il degrado personale e le anomalie mentali espresse sono caratteristiche, e il disadattamento sociale del paziente si verifica.
La malattia ha un decorso progressivo e i sintomi clinici dell'epilessia variano a seconda dello stadio di sviluppo dell'epilessia (prevalenza del focus epilettico).
Complicazioni e conseguenze
Anche nei casi lievi di epilessia focale con singole convulsioni rare, le fibre nervose sono danneggiate. La malattia ha una progressione progressiva, quando un attacco aumenta la probabilità del prossimo e l'area del danno cerebrale si espande.
I parossismi frequenti generalizzati agiscono distruttivamente sul tessuto cerebrale, possono svilupparsi in un epistato con un'alta probabilità di un esito letale. C'è anche il pericolo di edema cerebrale.
Le complicazioni e le conseguenze dipendono dal grado di distruzione delle strutture cerebrali, la gravità e la frequenza delle crisi epilettiche, comorbidità, la presenza di cattive abitudini, l'età, l'adeguatezza della politica adottata, trattamento e misure di riabilitazione, atteggiamento responsabile al trattamento del paziente.
A qualsiasi età, durante le cadute possono verificarsi lesioni di diversa gravità. L'ipersalivazione e la tendenza a vomitare i riflessi durante le crisi aumentano il rischio di ottenere sostanze liquide nel sistema respiratorio e lo sviluppo della polmonite da aspirazione.
Nell'infanzia, ha luogo l'instabilità dello sviluppo mentale e fisico. Le capacità cognitive spesso soffrono.
Lo stato psico-emotivo non è stabile - i bambini sono irritabili, capricciosi, spesso aggressivi o apatici, mancano di autocontrollo, non si adattano bene alla squadra.
Negli adulti, questi rischi sono integrati da traumatismi nell'esecuzione di lavori che richiedono maggiore attenzione. Durante le crisi, c'è un morso della lingua o della guancia.
Negli epilettici aumenta la probabilità di sviluppare depressione, disturbi mentali, disadattamento sociale. Le persone che soffrono di epilessia sono limitate nell'attività fisica, nella scelta della professione.
Diagnostica epilessia crittogena
Nella diagnosi dell'epilessia vengono usati molti metodi diversi per aiutare a differenziare questa malattia da altre patologie neurologiche.
Prima di tutto, il medico deve ascoltare le lamentele del paziente o dei suoi genitori, se è un bambino. Storia Compilato della malattia - Dettagli specificità flusso manifestazione (la frequenza delle crisi epilettiche, sincope, convulsioni e altre sfumature dei personaggi), durata della malattia, la presenza di tali malattie nei parenti del paziente. Questo sondaggio suggerisce un tipo di epilessia e localizzazione del focus epilettico.
I test del sangue e delle urine vengono assegnati per valutare le condizioni generali del corpo, la presenza di fattori come infezioni, intossicazioni, disturbi biochimici, per determinare la presenza di mutazioni genetiche nel paziente.
Viene eseguito un test neuropsicologico, che consente di valutare le capacità cognitive e lo stato emotivo. Il monitoraggio periodico consente di valutare l'impatto della malattia sul sistema nervoso e sulla psiche, inoltre, aiuta anche a stabilire il tipo di epilessia.
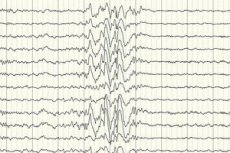
Ma soprattutto - uno strumento diagnostico con il quale siamo in grado di stimare l'intensità della attività elettrica del cervello (EEG), la presenza nei suoi dipartimenti di malformazioni vascolari, tumori, disturbi metabolici, etc ...
Elettroencefalografia (EEG) è il principale metodo di diagnosi poiché dimostra deviazioni dalla norma dell'intensità delle onde cerebrali anche attacco è - aumentato sequestro determinate porzioni o l'intero cervello. Modello EEG dell'epilessia parziale criptogenica - attività dell'onda di picco o di onde lente stabili in alcune parti del cervello. Con l'aiuto di questo studio sulle specifiche dell'elettroencefalogramma, è possibile stabilire il tipo di epilessia. Ad esempio, con la sindrome di Vest, si osservano onde lente aritmiche irregolari, quasi non sincronizzate con ampiezza anomala e scariche spike. Nella maggior parte dei casi la sindrome di Lennox-Gastaut elektroentsefallogramme al risveglio periodo rivelato generalizzata irregolare onda lenta attività picco con una frequenza di 1,5-2,5 Hz è spesso un'asimmetria ampiezza. Durante il riposo notturno per questa sindrome è caratterizzato dalla registrazione di scariche ritmiche rapide con una frequenza di circa 10 Hz.
Nel caso dell'epilessia criptogenetica, questo è l'unico modo per confermare la sua presenza. Sebbene ci siano casi in cui anche immediatamente dopo un attacco convulso su un elettroencefalogramma, i cambiamenti nella forma di onde cerebrali non sono registrati. Questo può essere un segno che i cambiamenti nell'attività elettrica avvengono nelle strutture profonde del cervello. I cambiamenti nell'elettroencefalogramma possono verificarsi anche in pazienti senza epilessia.
Sono usati metodi di neuroimaging per uso moderno: computer, risonanza, tomografia a emissione di positroni. Questa diagnosi strumentale consente di valutare i cambiamenti nella struttura della sostanza cerebrale dovuti a traumi, anomalie congenite, malattie, intossicazioni, rilevamenti di neoplasie, ecc. La tomografia ad emissione di positroni, chiamata anche risonanza magnetica funzionale, aiuta a identificare non solo i disordini strutturali, ma anche quelli funzionali.
Tasche più profonde di attività elettrica anormale consentono di identificare la tomografia computerizzata a emissione di un fotone, la spettroscopia di risonanza può rilevare anomalie nei processi biochimici nella sostanza cerebrale.
Il metodo di diagnosi sperimentale e non comune è un'encefalografia magnetica che rileva le onde magnetiche emesse dai neuroni del cervello. Ti permette di esplorare le strutture più profonde del cervello, inaccessibile all'elettroencefalografia.
Diagnosi differenziale
La diagnosi differenziale viene eseguita dopo i massimi studi possibili. La diagnosi di epilessia criptogenetica è determinata dal metodo di escludere altre specie e le cause delle crisi epilettiche, rivelate durante la diagnosi, così come la predisposizione ereditaria.
Non tutte le istituzioni mediche hanno lo stesso potenziale diagnostico, quindi questa diagnosi presuppone il proseguimento degli studi diagnostici ad un livello più alto.
Trattamento epilessia crittogena
Non esiste un unico metodo per il trattamento dell'epilessia, tuttavia sono stati sviluppati standard chiari che sono stati osservati per migliorare la qualità del trattamento e la vita dei pazienti.
Prevenzione
Poiché le cause di questo particolare tipo di epilessia non sono stabilite, le misure preventive hanno una direzione comune. Uno stile di vita sano è l'assenza di cattive abitudini, la piena nutrizione, l'attività motoria fornisce una buona immunità e previene lo sviluppo di infezioni.
Un'atteggiamento attento alla loro salute, un esame tempestivo e il trattamento di malattie e lesioni aumenta anche la probabilità di evitare questa malattia.
Previsione
L'epilessia criptogenetica si manifesta a qualsiasi età e non ha un complesso specifico di sintomi, ma si manifesta in vari modi: sono possibili diversi tipi di convulsioni e tipi di sindromi. Fino ad ora non esiste un unico metodo per la cura completa dell'epilessia, ma il trattamento antiepilettico aiuta nel 60-80% dei casi di tutti i tipi di malattie.
La durata media della malattia è di 10 anni, dopo i quali i sequestri possono fermarsi. Tuttavia, dal 20 al 40% dei pazienti soffre di epilessia per tutta la vita. Circa un terzo di tutti i pazienti con qualsiasi tipo di epilessia e muoiono per le cause associate ad esso.
Ad esempio, le forme criptogenetiche della sindrome di Vest sono prognosticamente sfavorevoli. Nella maggior parte dei casi, diventano la sindrome di Lennox-Gastaut, le cui forme lievi sono suscettibili al controllo della droga, mentre i pazienti generalizzati con convulsioni frequenti e gravi possono rimanere a vita e sono accompagnati da una grave degradazione intellettuale.
In generale, la prognosi dipende molto dal momento dell'inizio del trattamento, quando inizia nelle fasi iniziali - la previsione è più favorevole.
Il risultato dell'epilessia può essere una disabilità permanente. Se una persona sviluppa un persistente disturbo della salute a causa della malattia, portando a una restrizione delle attività della vita, allora questo è stabilito dalle competenze mediche e sociali. Decide anche sull'assegnazione di un gruppo specifico di disabilità. Per risolvere questo problema, prima di tutto, rivolgersi al medico curante, che presenterà il paziente alla commissione.