Esperto medico dell'articolo

Nuove pubblicazioni
Sindrome di Dejerine
Ultima recensione: 04.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
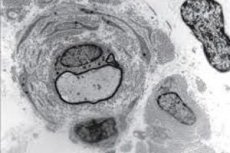
La sindrome di Dejerine è una malattia piuttosto rara, basata su una predisposizione genetica. È anche chiamata neuropatia ipertrofica. Si può affermare a prima vista che la malattia è incurabile, poiché tutte le malattie causate da mutazioni e alterazioni genetiche non possono essere curate.
La prima descrizione della malattia si deve al neurologo francese Dejerine, che inizialmente ipotizzò che avesse profonde radici genetiche. Notò che la malattia si trasmette di generazione in generazione, osservata all'interno di una stessa famiglia. Condusse anche studi sperimentali che gli permisero di concludere che i geni dominanti sono responsabili della trasmissione della malattia. Pertanto, tramite la consulenza genetica, è possibile prevedere in anticipo se un bambino nascerà sano o svilupperà la sindrome di Dejerine.
Purtroppo, non esiste alcun modo per prevenirne lo sviluppo. Se la malattia viene trasmessa al bambino, si svilupperà inevitabilmente.
 [ 1 ]
[ 1 ]
Epidemiologia
Esistono attualmente molti tipi di sindrome di Dejerine. Tuttavia, presentano tutte caratteristiche simili: si manifestano tra la nascita e i 7 anni. Allo stesso tempo, circa il 20% dei casi si manifesta nel primo anno di vita. Nel secondo anno di vita, la malattia si manifesta nel 16% dei casi.
La sindrome di Dejerine-Sottas è la più comune. Si riscontra in circa il 43% dei casi. In circa il 96% dei casi provoca una disabilità completa, costringendo la persona a usare una sedia a rotelle.
Al secondo posto si colloca la sindrome di Dejerine-Klumpke, che si verifica in circa il 31% dei casi. Al terzo posto si colloca la sindrome di Dejerine-Russo, la cui frequenza di insorgenza è di circa il 21%. Allo stesso tempo, la sindrome di Dejerine-Russo è caratterizzata dalla comparsa di sintomi stabili entro un anno nei pazienti che hanno subito un ictus o un altro ictus cerebrovascolare in forma acuta.
La sindrome dolorosa si sviluppa in modo irregolare. In circa il 50% dei pazienti, il dolore si manifesta entro 1 mese dall'ictus, nel 37% entro un periodo compreso tra 1 mese e 2 anni, e nell'11% dei casi dopo 2 anni. Parestesie e allodinia si riscontrano nel 71% dei pazienti.
Le cause Sindrome di Dejerine
La causa principale della sindrome di Dejerine è una mutazione genetica trasmessa con modalità autosomica. Tuttavia, numerosi fattori genetici possono influenzare lo sviluppo della patologia, colpendo la persona e il suo cervello. Le principali cause della malattia includono:
- traumi, lesioni e altri effetti negativi. Questo vale soprattutto per i nervi cranici. La malattia può anche essere una conseguenza di commozioni cerebrali;
- fratture delle ossa situate alla base del cranio;
- infiammazione delle meningi, che si manifesta in forma acuta. L'infiammazione può avere varie origini. Può essere causata da agenti infettivi, infiammazione, reazione allergica. Inoltre, lo sviluppo della sindrome può essere conseguenza di un trauma;
- infiammazione delle meningi di varia origine divenuta cronica;
- aumento della pressione intracranica.
Fattori di rischio
Esistono alcuni fattori di rischio che possono scatenare la malattia. Le persone esposte a questi fattori sono più suscettibili alla malattia rispetto ad altre. I fattori di rischio includono anche alcune malattie che accompagnano la patologia.
Il gruppo a rischio include i pazienti affetti da tumori cerebrali. Un tumore che esercita una pressione sul midollo allungato può essere considerato un fattore di rischio. Questo gruppo include anche vari tipi di tubercolomi, danni vascolari e sarcoidosi. Il danno cerebrale si verifica a causa della pressione esercitata sul cervello. I danni ai vasi cerebrali possono essere di diversa natura. In primo luogo, si tratta di lesioni emorragiche, embolie, trombosi, aneurismi e malformazioni.
Inoltre, uno dei fattori che contribuiscono allo sviluppo della sindrome di Dejerine sono malattie concomitanti come poliencefalite, sclerosi multipla e poliomielite. Il rischio può essere rappresentato anche da quelle malattie che sono accompagnate da un'interruzione del normale funzionamento del cervello, un'interruzione del suo apporto di sangue. Innanzitutto, bisogna prestare attenzione alle interruzioni del flusso sanguigno nel letto arterioso. Il gruppo a rischio include anche i pazienti suscettibili all'interruzione dell'apporto di sangue al dodicesimo nervo, al suo nucleo, all'ansa mediale e alla piramide.
Anche la siringobulbia e la paralisi bulbare contribuiscono allo sviluppo della malattia. Questi fattori rappresentano un rischio elevato, poiché sono caratterizzati da una progressione costante.
Anche i tumori cerebellari di vario tipo possono essere considerati un fattore di rischio.
I pazienti con anomalie congenite del cervello rientrano nei gruppi a rischio. Se una persona con tale anomalia è esposta ad agenti infettivi, tossici e degenerativi, il rischio di sviluppare la malattia aumenta significativamente. Fattori come sostanze chimiche caustiche e radioattive possono provocare lo sviluppo della patologia. Possono causare una mutazione genetica. Pertanto, le donne esposte a sostanze chimiche tossiche, così come quelle che vivono in una zona ad alto tasso di radiazioni, possono rientrare nel gruppo a rischio. In questo caso, la predisposizione alla malattia aumenta notevolmente.
Patogenesi
La patogenesi della malattia è causata da una mutazione genetica. Essa contribuisce alla distruzione della struttura delle guaine dei nervi del tronco. Con lo sviluppo della malattia, si osserva una crescita eccessiva delle guaine connettive, che fanno parte del tessuto nervoso. Di conseguenza, il tessuto connettivo si ipertrofizza e si deposita sostanza mucosa tra le connessioni nervose. Ciò porta a un significativo ispessimento dei tronchi nervosi, delle radici spinali e dei tratti cerebellari. La loro forma cambia. I processi degenerativi colpiscono il tessuto nervoso e i nervi spinali.
Sintomi Sindrome di Dejerine
La sindrome di Dejerine può manifestarsi in modi completamente diversi. È importante comprendere che esistono molte varianti di questa malattia, e ciascuna di esse si manifesta con sintomi completamente diversi. Pertanto, è opportuno analizzare i sintomi caratteristici di ogni singola tipologia di questa sindrome.
Tuttavia, esistono diversi segnali precoci che possono generalmente indicare la probabilità che un bambino sviluppi una patologia. Nelle fasi iniziali, diverse tipologie possono presentare molte caratteristiche simili.
Primi segni
Nella maggior parte dei casi, la malattia si manifesta già in età prescolare. Tuttavia, i suoi primi segni possono essere sospettati quasi dalla nascita del bambino. Se il bambino si sviluppa più lentamente rispetto ai suoi coetanei, questo può essere il primo segnale d'allarme. È necessario prestare particolare attenzione al bambino che non si siede all'età appropriata, fa il primo passo in ritardo o inizia a muoversi autonomamente.
Anche l'aspetto del bambino può rivelare molto. Di solito, i muscoli facciali del bambino sono cadenti. Braccia e gambe iniziano gradualmente a deformarsi. Diventano meno sensibili, praticamente non reagiscono a nulla. Questa condizione può peggiorare costantemente, fino all'atrofia muscolare.
Non appena il bambino inizia a svilupparsi in modo anomalo, è necessario consultare un medico. È necessaria una consulenza con un neurologo.
Durante l'esame obiettivo, il medico rileva ulteriori segni indicativi della sindrome. Si osservano contrazioni fibrillari dei muscoli. Molti riflessi tendinei non sono evidenti. Le pupille possono essere contratte e nella maggior parte dei casi non reagiscono alla luce. Il medico conferma i segni di indebolimento dei muscoli facciali.
Fasi
Esistono stadi lievi (iniziali), stadi moderati e stadi gravi. Lo stadio iniziale è quello in cui compaiono i primi segni della malattia. Questo stadio si verifica solitamente durante l'infanzia.
La fase intermedia è caratterizzata da un ritardo marcato nello sviluppo motorio e del linguaggio, vari disturbi motori, sensibilità compromessa, perdita di alcuni riflessi e reazioni visive compromesse.
Stadio grave: sordità neurosensoriale, deformità scheletriche, disturbi del tono muscolare, nistagmo. Progressione della malattia. Sfocia in disabilità.
Forme
Esistono numerose varianti della sindrome di Dejerine, a seconda del tipo e della gravità della lesione. Le più comuni sono la sindrome alternante, la sindrome di Dejerine-Sotta, la sindrome di Dejerine-Klumpke e la sindrome di Dejerine-Rousset.
[ 21 ]
Sindrome alternante di Dejerine
Se un bambino è affetto da sindrome alternante, la lingua è la prima a essere paralizzata. Inoltre, non è colpita l'intera lingua, ma solo una parte. L'emiparesi si sviluppa sul lato opposto. La sensibilità alle vibrazioni raggiunge strati profondi. Il bambino praticamente non distingue le sensazioni tattili. La causa è una trombosi o un'occlusione dell'arteria basilare. Questo è ciò che interrompe l'innervazione e l'afflusso di sangue al midollo allungato.
Sindrome di Dejerine Klumpke
Nella sindrome di Dejerine-Klumpke, i rami inferiori dell'articolazione della spalla sono paralizzati. Non l'intero arto è paralizzato, ma solo una parte di esso. Si sviluppano gradualmente paresi e paralisi delle mani. La sensibilità delle aree corrispondenti è notevolmente ridotta. Le condizioni dei vasi sanguigni cambiano. Le reazioni pupillari sono anomale.
La paralisi si diffonde gradualmente agli strati profondi della struttura muscolare. Si osserva un grave intorpidimento. Prima si intorpidiscono le mani, poi gli avambracci e i gomiti. Nei casi più gravi, può essere colpito anche il nervo toracico. Si sviluppano anche numerose ptosi e miosi.
Sindrome di Dejerine-Roussy
Questa sindrome è caratterizzata da danni alle arterie perforanti. Sono danneggiate anche le aree circostanti l'arteria e le aree cerebrali innervate dall'arteria interessata. Questa sindrome è anche chiamata sindrome del dolore cronico o sindrome del dolore talamico (post-ictus).
Il nome è dovuto al fatto che la sindrome è accompagnata da un dolore intenso, costante e lancinante. Il dolore è spesso insopportabile. La malattia è inoltre accompagnata da una sensazione di indolenzimento e torsione di tutto il corpo. Si osserva anche iperpatia, a causa della quale alcuni muscoli assumono un tono eccessivo. Tuttavia, la sensibilità è notevolmente ridotta. La malattia è inoltre caratterizzata da attacchi di panico, pianto innaturale, urla o risate.
In questo caso, il danno è prevalentemente limitato a un lato, che potrebbe essere una gamba o un braccio. Nelle zone colpite si osservano principalmente dolore intenso e sensazione di bruciore. Il dolore sfinisce il paziente. Può essere aggravato da diversi fattori. Il dolore può essere aggravato da emozioni sia positive che negative. Il dolore può anche essere aggravato dal caldo, dal freddo e da vari movimenti.
Spesso la malattia è difficile da differenziare, da separare da altre patologie. Presenta molti segni simili ad altre lesioni nevralgiche. A volte può essere diagnosticata definitivamente solo dopo che la sindrome dolorosa si è completamente formata.
Sindrome di Dejerine Sottas
La sindrome di Dejerine-Sotta è una malattia genetica. Nel corso di questa malattia, lo spessore dei nervi del tronco è compromesso. La malattia può essere diagnosticata nelle prime fasi della gravidanza tramite consulenza genetica. Alla nascita, il bambino non è diverso da un bambino sano. Poi, crescendo e sviluppandosi, si nota che lo sviluppo del bambino è molto lento. Movimenti scarsi, il linguaggio non si sviluppa. I muscoli sono molto rilassati, il bambino non è in grado di sostenere la testa, il collo e il corpo. Le reazioni visive sono compromesse. Il bambino è molto indietro rispetto ai suoi coetanei nello sviluppo. La sensibilità diminuisce progressivamente, i muscoli si atrofizzano gradualmente. Lo sviluppo completo non si verifica. Gradualmente, l'atrofia si trasmette al sistema scheletrico, portando alla disabilità.
Sindrome di Neri Dejerine
Nella sindrome di Neri Dejerine, le radici posteriori del midollo spinale sono costantemente irritate. La causa è l'osteocondrosi, ovvero vari tumori che colpiscono il cervello e premono su di esso. Anche ernie, schiacciamenti e lesioni contribuiscono alla pressione sulle radici. Inoltre, questo può verificarsi a causa di forti escrescenze ossee. La manifestazione principale è un forte dolore nel punto in cui si verifica la pressione sul cervello e sulle sue radici.
Nella maggior parte dei casi, questa sindrome non è la principale, ma concomitante a diverse altre patologie e disturbi. Ad esempio, tradizionalmente accompagna l'osteocondrosi. Una caratteristica distintiva è un dolore acuto nella regione lombare e un dolore persistente al collo e alla testa, che impedisce di sollevare completamente la testa da una posizione sdraiata. Gradualmente, questa zona si indurisce e la sensibilità si perde gradualmente. Si osservano anche spasmi muscolari. Gradualmente, gli arti subiscono alterazioni patologiche.
Sindrome di Landouzy Dejerine
Un sinonimo è miopatia. Il nome della malattia indica un indebolimento muscolare in continua progressione. Parallelamente, si osserva lo sviluppo di diverse patologie muscolari, tra cui processi distrofici. Possiamo dire che non si tratta di una malattia a sé stante, ma di un intero gruppo di patologie. Sono colpite la spalla, la scapola e il lato facciale. La malattia è una patologia genetica, trasmessa di generazione in generazione.
Si sviluppa in diverse fasi. Nella prima fase, si sviluppa debolezza facciale, a causa della quale i muscoli facciali non solo si indeboliscono, ma perdono anche forma e si deformano. Di conseguenza, il viso acquisisce lineamenti irregolari e distorti. Il più delle volte, la malattia è riconoscibile da una bocca arrotondata e da labbra inferiori e superiori cadenti.
Gradualmente, la malattia progredisce a tal punto che la persona non riesce più a chiudere la bocca. La tiene aperta prima durante il sonno, poi anche durante il giorno. Gradualmente, la debolezza muscolare colpisce i muscoli del cingolo scapolare.
In rari casi, i muscoli faringei e la lingua possono indebolirsi. Tuttavia, questo sintomo non ha valore diagnostico e non è così pronunciato come altri sintomi.
Nella fase più grave, la persona sviluppa debolezza dei muscoli scheletrici. Prima si indeboliscono le braccia, poi le gambe. La prognosi è deludente: disabilità.
Diagnostica Sindrome di Dejerine
La sindrome di Dejerine può essere diagnosticata in base ai sintomi e alle manifestazioni cliniche caratteristiche della malattia. In alcuni casi, il quadro clinico è così pronunciato che la malattia può essere sospettata anche solo sulla base di un esame obiettivo. Ma in realtà, la situazione è molto più complessa. Altre malattie neurologiche possono manifestarsi in modo simile. Pertanto, è importante analizzare immediatamente i segni clinici presenti, analizzando i dati dell'esame soggettivo e oggettivo. La conclusione finale si basa su esami di laboratorio e strumentali. È inoltre necessario studiare l'anamnesi e la storia familiare.
Test
La diagnosi di sindrome di Dejerine può essere confermata tramite l'analisi del liquido cerebrospinale e la biopsia. L'esame del liquido cerebrospinale consente di rilevare un gran numero di proteine e frammenti proteici. Questi rappresentano il segno distintivo della sindrome di Dejerine.
In alcuni casi, questo è sufficiente per stabilire una diagnosi precisa. Ma a volte sorgono dubbi. Ad esempio, la presenza di pochi frammenti proteici nel liquido cerebrospinale potrebbe indicare altre patologie neurologiche. Pertanto, in questi casi dubbi, viene eseguita una biopsia. La biopsia richiede il prelievo di fibre nervose, prelevate dai muscoli del gomito e del polpaccio. La sindrome di Dejerine è indicata con precisione dalla presenza di ipertrofia del tessuto nervoso. In questa patologia, le guaine delle fibre nervose si ispessiscono notevolmente.
Inoltre, studiando i tessuti nervosi al microscopio, si può stabilire che non solo le membrane si ispessiscono, ma anche il numero di fibre stesse si riduce significativamente. Si verifica anche una demineralizzazione. Il numero di fibre nervose si riduce.
Diagnostica strumentale
A volte, tuttavia, anche i test non sono sufficienti per accertare con certezza la presenza della sindrome di Dejerine. In tal caso, è necessaria un'apparecchiatura specifica. Nella maggior parte dei casi, vengono utilizzate la risonanza magnetica e la computer imaging. Questi metodi consentono di rilevare il grado di danno al dodicesimo nervo. Dopo aver ricevuto i risultati, il medico seleziona il trattamento più adatto. La terapia viene prescritta da un neurologo o un neurochirurgo.
Diagnosi differenziale
L'essenza della diagnosi differenziale è distinguere chiaramente i sintomi di una malattia da quelli di un'altra che presenta manifestazioni simili. Nella sindrome di Dejerine, questo è molto importante, poiché da questo dipendono la prognosi e il trattamento successivi. Questa malattia può spesso essere confusa con altre patologie neurologiche, come la paralisi.
Una volta confermata la diagnosi, è necessario differenziarla da altre varianti, ovvero determinare con certezza a quale tipo specifico di sindrome di Dejerine si ha a che fare. Una biopsia e l'analisi del liquido cerebrospinale possono essere d'aiuto.
La presenza della sindrome di Dejerine è indicata dalla presenza di proteine nel liquido cerebrospinale e da fibre nervose ispessite in una biopsia. Il tipo di sindrome è solitamente determinato dal quadro clinico e dai segni patognomonici, ovvero dai segni caratteristici di una particolare malattia, unici per essa, e che ne costituiscono la caratteristica distintiva.
Chi contattare?
Trattamento Sindrome di Dejerine
Poiché la malattia è genetica, è importante comprendere che sarà impossibile curarla completamente e liberarsene. Molto probabilmente, la malattia progredirà e non sarà possibile fermarla in alcun modo. Ma questo non significa che non valga la pena curarla. È sufficiente scegliere il trattamento con la massima attenzione e razionalità. Può rallentare la progressione della malattia e alleviare significativamente la sofferenza del paziente.
Nella scelta del trattamento, si è guidati dal fatto che la terapia eziologica è irrazionale. La terapia eziologica è intesa come un trattamento volto a eliminare la causa. Per una malattia genetica, è impossibile eliminare la causa. Pertanto, rimane la terapia sintomatica, ovvero una terapia volta ad alleviare i sintomi della malattia, migliorare le condizioni generali e il benessere del paziente. I regimi di trattamento possono essere completamente diversi. Tutto dipende da quale sintomo predomina, da ciò che preoccupa il paziente in quel momento. Per garantire il sollievo dai sintomi e allo stesso tempo prevenire l'ulteriore progressione della malattia, si utilizza una terapia complessa.
La terapia complessa di solito include antidolorifici, poiché la sindrome è quasi sempre accompagnata da sensazioni dolorose. In assenza di dolore (il che accade piuttosto raramente), tali farmaci possono essere esclusi.
È fondamentale la terapia metabolica, che migliora i processi metabolici, promuove una migliore nutrizione dei tessuti e rimuove i metaboliti. Questo tipo di trattamento è principalmente mirato al mantenimento del tessuto muscolare, poiché è molto suscettibile a processi degenerativi e può successivamente atrofizzarsi. L'obiettivo principale di questa terapia è prevenire l'atrofia.
È inoltre necessario utilizzare farmaci per migliorare la conduttività nervosa. Questi consentono di normalizzare i processi metabolici nel tessuto nervoso, ripristinando o mantenendo la sensibilità delle terminazioni nervose e prevenendo la morte dei recettori.
Oltre al trattamento farmacologico, potrebbe essere prescritta anche la fisioterapia. Potrebbe essere necessario un ciclo di massaggi, terapia manuale e diverse terapie alternative. Attualmente, esistono molti prodotti ortopedici che permettono di prevenire lo sviluppo di patologie scheletriche. È anche possibile prevenire lo sviluppo di deformità del piede. Anche le contratture articolari possono essere prevenute con l'aiuto di prodotti ortopedici.
A volte il trattamento può essere mirato a eliminare la causa che ha provocato la malattia. Naturalmente, se non si tratta di una causa genetica. Ad esempio, in alcuni casi, sebbene una persona abbia una predisposizione genetica, la malattia non si manifesta. Ma poi, a causa di qualche fattore, la malattia inizia a svilupparsi o progredire. Pertanto, la causa può essere una trombosi arteriosa. L'arteria danneggiata comprime la parte adiacente del cervello, interrompendone l'afflusso di sangue. In questo caso, è consigliabile cercare di eliminare la causa, ovvero rimuovere la trombosi. In questo caso, potrebbe essere necessario un intervento chirurgico.
In altri casi è necessaria una terapia di supporto continuativa.
Medicinali
I farmaci vengono utilizzati esclusivamente per eliminare i sintomi. Ad esempio, per trattare la sindrome del dolore, si raccomanda l'uso di cabrazepam alla dose di 3-5 mg/kg di peso corporeo 2-3 volte al giorno.
Il ketorolac può anche essere raccomandato alla concentrazione di 60 mg/die, 2 volte al giorno. Il trometamolo viene utilizzato alla dose di 60 mg/die, 2 volte al giorno, mentre il chetone viene utilizzato alla dose di 50 mg, 1-2 volte al giorno; la dose massima giornaliera è di 100-150 mg.
Vitamine
Le vitamine sono necessarie per mantenere le condizioni generali dell'organismo, normalizzare il sistema immunitario e proteggere da malattie infettive e di altro tipo. Le vitamine contribuiscono anche a migliorare il benessere generale del bambino. Le principali vitamine necessarie per il normale funzionamento dell'organismo (dose giornaliera):
- B – 2-3 mg
- PP – 30 mg
- H – 7-8 mcg
- C – 250 mg
- D – 20 mcg
- E – 20 mg.
Trattamento fisioterapico
La fisioterapia non può curare la sindrome di Dejerine. Tuttavia, in alcuni casi, può essere utilizzata. Di solito, viene utilizzata per uno scopo specifico. Pertanto, l'elettroforesi aumenta significativamente la permeabilità tissutale e può essere utilizzata per garantire che i farmaci penetrino nei tessuti in modo più rapido ed efficace. Alcune procedure fisioterapiche possono ridurre il dolore, rilassare i muscoli e alleviare gli spasmi. Possono migliorare significativamente le condizioni generali del corpo. La fisioterapia aiuta anche ad alleviare il dolore.
Rimedi popolari
Esistono rimedi popolari che aiutano ad alleviare il disagio del paziente. È impossibile curare la sindrome di Dejerine, ma questo non significa che si debba arrendersi e non impegnarsi per alleviare o ridurre i sintomi. I rimedi popolari aiutano a superare i sintomi e ad alleviare il dolore. Forniscono un significativo supporto psicologico al paziente. La malattia non può essere curata, ma è possibile rallentarne la progressione.
In caso di paresi, paralisi e indebolimento dei muscoli facciali, si consiglia l'uso di avena. Utilizzare succo di avena verde. Assumere un terzo di bicchiere due volte al giorno. È preferibile assumerlo prima dei pasti. L'effetto è un rafforzamento generale.
Paralisi e paresi sono alleviate dall'uso di menta e melissa. Si consiglia di prepararle in infusione, preparare un decotto e berle calde. Queste erbe sono relativamente sicure, quindi possono essere utilizzate in grandi quantità, ma non in modo incontrollato. È consentito circa un litro al giorno. Queste erbe possono anche essere aggiunte a una tisana a piacere. Bevetele in quantità illimitate, a seconda del gusto e dell'umore. Di solito permettono ai muscoli di rilassarsi e normalizzano anche lo stato del sistema nervoso, avendo un effetto calmante.
Menta e melissa possono anche essere mescolate con il vischio, in proporzioni approssimativamente uguali, e utilizzate come decotto. In questo caso, si consiglia di utilizzarle in quantità limitate, circa 20-30 ml tre volte al giorno. Questo infuso aiuta a eliminare spasmi e dolori, a rilassare i muscoli e a calmare il sistema nervoso.
I bagni con erbe medicinali hanno un effetto benefico sul corpo. Si può preparare un bagno con questa miscela. Per farlo, si preparano separatamente circa 2-3 litri di infuso forte, che poi si versa in una vasca da bagno a temperatura confortevole. Si consiglia di fare bagni per 20-30 minuti. Permettono di tonificare i muscoli e normalizzare l'attività del sistema nervoso. Le erbe possono essere combinate e alternate. Si può usare un decotto di conifere: pino, abete, cedro. Si possono aggiungere camomilla, tiglio, lampone, ortica.
[ 31 ], [ 32 ], [ 33 ], [ 34 ]
Trattamento a base di erbe
In caso di problemi di coordinazione, paresi e paralisi, si può ricorrere all'efedra. Si assume sotto forma di decotto. Si preparano circa 5 g di efedra in 500 ml di acqua bollente. Si possono bere 2-3 cucchiai tre volte al giorno.
Inoltre, per normalizzare la condizione muscolare, calmare, alleviare spasmi e dolori, si può utilizzare un decotto o un infuso di valeriana. La tintura alcolica di valeriana è disponibile in commercio. Il metodo di somministrazione è solitamente indicato sulla confezione.
Per preparare un decotto a casa, versate circa 5 g di erba in un bicchiere d'acqua e bevete il decotto durante il giorno. Potete anche aggiungerlo al tè.
Il decotto di camomilla può essere usato in modo simile. Ha inoltre un effetto antinfiammatorio, normalizza il sistema immunitario e il metabolismo.
Si consiglia di usare un decotto di calendula, 1 cucchiaio tre volte al giorno. Ha un effetto antinfiammatorio e allevia il gonfiore.
Omeopatia
Anche i rimedi omeopatici possono avere un effetto positivo, migliorare le condizioni generali dell'organismo ed eliminare singoli sintomi. Gli effetti collaterali sono rari se il dosaggio e le modalità di somministrazione vengono seguiti correttamente. È importante considerare che molte sostanze hanno un effetto cumulativo, il che significa che l'effetto si manifesterà solo al termine dell'intero ciclo di trattamento o dopo un certo periodo di tempo. È necessario osservare le precauzioni di base: consultare un medico prima dell'assunzione, poiché alcune sostanze potrebbero non essere compatibili tra loro o con i farmaci. Le conseguenze possono essere imprevedibili.
Per paralisi flaccida, paresi, ipercinesia e indebolimento dei muscoli facciali, si consiglia l'assunzione di Securinega sibirica. Si versano circa 15 g di foglie e rametti tritati in 250 ml di acqua bollente. Lasciare in infusione fino a raffreddamento. Filtrare e assumere un terzo di bicchiere, due volte al giorno.
- Raccolta n. 1. Per lesioni del midollo allungato, cervelletto
Prendi foglie di lampone, foglie di ribes, valeriana e erba cardiaca in un rapporto di 2:1:2:1. Assumi sotto forma di infuso, un terzo di bicchiere tre volte al giorno.
- Raccolta n. 2. Per paralisi spastica
Prendi le foglie di melissa, camomilla, salvia e menta in un rapporto di 1:1:2:2. Assumi sotto forma di infuso tre volte al giorno, un terzo di bicchiere.
- Raccolta n. 3. Per sindrome dolorosa, spasmi, paralisi
Prendi foglie di stevia, ortica, coni di luppolo comune e fiori di echinacea in un rapporto di 2:1:1:1. Assumi come infuso tre volte al giorno.
Trattamento chirurgico
In alcuni casi, solo l'intervento chirurgico può essere d'aiuto. Ad esempio, se il paziente ha una lesione o un tumore, questi devono essere rimossi. Anche la trombosi o l'occlusione delle arterie possono richiedere un intervento chirurgico.
In caso di patologie dei vasi sanguigni, è efficace l'intervento chirurgico intravascolare mininvasivo.
Se è interessata un'arteria specifica, potrebbe essere necessario un intervento chirurgico per migliorare la circolazione cerebrale e normalizzare l'innervazione di quest'area.
Ma in alcuni casi ci sono patologie che non possono essere operate. Possono essere varie anomalie congenite o lesioni.
Previsione
Il decorso della malattia è sempre progressivo, i periodi di remissione sono brevi. La prognosi è sfavorevole. Ciò è dovuto principalmente al fatto che i principali processi degenerativi si verificano nel sistema nervoso, il cervello. Con il progredire della malattia, la capacità lavorativa si perde. Infine, il paziente è costretto su una sedia a rotelle o a letto.
[ 39 ]