Esperto medico dell'articolo

Nuove pubblicazioni
Sindrome di Tolosa-Hunt
Ultima recensione: 04.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
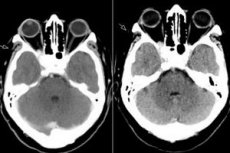
Sindrome della fessura orbitaria superiore, oftalmoplegia patologica: tutto questo non è altro che la sindrome di Tolosa-Hunt, ovvero una lesione delle strutture della fessura orbitaria superiore. Il processo solitamente coinvolge i vasi orbitari (arteriosi e venosi), le fibre nervose (nervi oculomotore, trocleare, abducente, nonché il primo ramo del nervo trigemino) e il seno cavernoso adiacente. La malattia può essere classificata come una patologia relativamente rara e piuttosto difficile da diagnosticare. [ 1 ]
Epidemiologia
La sindrome di Tolos-Hunt è stata descritta non molto tempo fa: circa 70 anni fa. Fu studiata dal neurologo spagnolo E. Tolos. Pochi anni dopo, il lavoro fu integrato dall'oculista inglese W. Hunt. I nomi dei medici-ricercatori divennero la base per il nome della sindrome.
La sindrome di Tolosa Hunt si riscontra in egual misura negli uomini e nelle donne. La patologia è solitamente monolaterale e si osserva con la stessa frequenza sul lato sinistro o destro. La sindrome bilaterale è possibile, ma si verifica solo in casi isolati.
L'età media dei soggetti colpiti è di 50 anni. In generale, la sindrome di Tolosa-Hunt può essere diagnosticata tra i 15 e gli 85 anni. La maggior parte dei pazienti appartiene alla fascia d'età anziana: lo sviluppo della malattia è facilitato da molteplici disturbi cardiovascolari, nonché da alterazioni tissutali legate all'età.
Il sintomo più comune della malattia è la manifestazione di un classico attacco di emicrania: il paziente avverte un improvviso mal di testa pulsante da un lato, "lancinante" o "torcente", con irradiazione alla cavità oculare. Poiché la sindrome di Tolosa Hunt non presenta sintomi specifici tipici, la patologia viene spesso definita un "camaleonte neurologico": la diagnosi è complessa e richiede la differenziazione da molte altre patologie.
I pazienti con sindrome di Tolosa Hunt si riscontrano periodicamente in diversi paesi del mondo, senza alcuna caratteristica territoriale o stagionale. Il tasso di incidenza è di 0,3-1,5 casi per 1 milione di abitanti. [ 2 ]
Le cause Sindrome di Tolosa-Hunt
Nel corso delle indagini sulle cause dello sviluppo della sindrome di Tolosa Hunt, gli scienziati hanno scoperto i seguenti fatti:
- nella maggior parte dei casi la malattia è provocata dall'infiammazione immunitaria della parete esterna del seno cavernoso;
- in alcuni casi le cause erano malformazioni vascolari, processi tumorali nel cervello (forme primarie e secondarie), pachimeningite cranica localizzata, miosite orbitaria, periarterite nodosa e formazione di trombi nel seno cavernoso;
- In circa il 30% dei pazienti non è possibile determinare la causa del disturbo, per cui è stata formulata la diagnosi di sindrome di Tolosa Hunt idiopatica.
Consideriamo più in dettaglio queste presunte ragioni.
- Lo sviluppo autoimmune della sindrome è associato sia all'ipotermia che a recenti patologie infettive, nonché a stress profondo. La forma autoimmune della malattia è caratterizzata da: esordio acuto, decorso ricorrente, elevata efficacia della terapia con glucocorticoidi. Questa forma della malattia colpisce più frequentemente gli uomini.
- Le malformazioni vascolari si verificano spesso nell'ipertensione arteriosa scompensata. Le donne sono più frequentemente colpite. La malattia esordisce in modo acuto, il dolore è moderato, praticamente senza esoftalmo o chemosi.
- Tra i processi tumorali in grado di portare allo sviluppo della sindrome di Tolosa Hunt, i più comuni sono i tumori cerebrali primari, i tumori metastatici con focolai primari nei polmoni, nei bronchi, nella prostata o le metastasi del melanoma cutaneo.
- La pachimeningite cranica localizzata causa un esordio acuto della sindrome in assenza di segni cerebrali e meningei generali, senza esoftalmo. La diagnosi è confermata morfologicamente durante la biopsia.
- La miosite orbitaria ha un esordio subacuto, con dolore intenso ed esoftalmo, chemosi pronunciata e visione doppia.
- La trombosi del seno cavernoso causa oftalmoplegia totale. La diagnosi è confermata dalla risonanza magnetica.
- La periarterite nodulare può causare lo sviluppo della sindrome di Tolosa Hunt diversi mesi dopo l'insorgenza della malattia.
Nella maggior parte dei casi, il meccanismo autoimmune è alla base della formazione della patologia, come dimostrato da molti specialisti. La natura autoimmune è indicata, in particolare, dai seguenti fattori:
- corso ricorrente;
- disturbi dismunici;
- dissociazione proteina-cellula nel liquido cerebrospinale e aumento dei livelli di citochine proinfiammatorie nel liquido cerebrospinale e nel siero sanguigno. [ 3 ]
Fattori di rischio
Gli scienziati non hanno ancora determinato la causa esatta della sindrome di Tolosa Hunt. Tuttavia, sono riusciti a identificare alcuni fattori che influenzano lo sviluppo di questo disturbo:
- Predisposizione genetica alle malattie autoimmuni in generale. Se un membro della famiglia ha sofferto o soffre di una malattia autoimmune, anche altri parenti potrebbero presentare patologie simili o con meccanismi di sviluppo simili. Questo fattore è ancora un'ipotesi che richiede ulteriori ricerche e prove.
- Fattori ambientali, tra cui abitudini alimentari, condizioni ambientali, qualità dell'acqua, rischi industriali, ecc.
- Situazioni di forte stress, stress frequente e shock psico-emotivi, forti cambiamenti ormonali (tra cui gravidanza, menopausa, ecc.).
- Malattie infettive croniche di lunga durata, tra cui epatite, infezione da herpesvirus, citomegalovirus, ecc.
- Ipotermia, radiazioni, altri forti fattori irritanti e dannosi.
Patogenesi
Il meccanismo eziologico dello sviluppo della sindrome di Tolosa Hunt non è stato completamente chiarito. Il ruolo determinante è attribuito alle reazioni autoimmuni. Molti scienziati presumono che le infezioni virali e microbiche, le situazioni di stress e le radiazioni agiscano solo come fattori scatenanti. Non vi sono prove concrete della relazione tra l'ingresso di microrganismi patogeni nell'organismo e lo sviluppo della sindrome di Tolosa Hunt. Tuttavia, vi sono sospetti sul coinvolgimento del citomegalovirus nel processo autoimmune, che contribuisce alla formazione di granulomi. [ 4 ]
Lo schema patogenetico è causato dalla comparsa di un processo infiammatorio granulomatoso locale nell'area della parete esterna del seno cavernoso, nel tratto infraclinoideo o sopraclinoideo dell'arteria carotide interna, che ne determina il restringimento. Un ruolo importante è svolto anche dal disordine della protezione immunitaria umorale e cellulare. Il versante umorale della sindrome è associato a un'aumentata formazione di anticorpi citoplasmatici anti-neutrofili che agiscono contro gli enzimi proteinasi-3, mieloperossidasi e una specifica proteina di membrana in grado di legare le endotossine. Presumibilmente, gli anticorpi citoplasmatici stimolano i neutrofili esistenti, attaccando così gli organi "bersaglio"; in particolare, il processo infiammatorio si sviluppa nella parete esterna del seno cavernoso.
Anche i cambiamenti cellulari giocano un ruolo nello sviluppo della sindrome di Tolosa Hunt. Ciò è dimostrato dalla predominanza di linfociti T, macrofagi e plasmacellule nei granulomi.
Sono disponibili informazioni su strutture endoteliali altamente attive e citochine antinfiammatorie, il che indica una tendenza del processo patologico a diventare cronico.
In casi isolati sono state notate alterazioni necrotiche focali nell'area della parete esterna del seno cavernoso.
Sintomi Sindrome di Tolosa-Hunt
I sintomi caratteristici della sindrome di Tolosa Hunt si manifestano improvvisamente e inaspettatamente nel paziente. I sintomi principali sono considerati i seguenti:
- Dolore intenso nella zona della cavità oculare, estremamente fastidioso, perforante, che si diffonde dalla zona frontale alle arcate sopraccigliari, agli occhi e poi a tutta la testa.
- Visione doppia, che si manifesta dopo l'insorgenza del dolore. Diventa estremamente difficile per una persona concentrarsi visivamente ed esaminare qualsiasi oggetto.
- Il disturbo della funzione motoria del bulbo oculare, o cosiddetta oftalmoplegia, è prevalentemente monolaterale. Può manifestarsi in gradi diversi, a seconda della gravità del processo patologico e dell'estensione della lesione.
- Edema congiuntivale.
- Spostamento anteriore del bulbo oculare (esoftalmo, occhi “sporgenti”).
- Deviazione laterale dell'asse visivo di un bulbo oculare, strabismo, tipico del danno nervoso monolaterale.
- Peggioramento generale della salute, leggero aumento della temperatura, debolezza, irritabilità.
Il quadro clinico progredisce gradualmente, i sintomi cambiano e peggiorano, ma possono scomparire all'improvviso così come sono comparsi. Tuttavia, in assenza della terapia necessaria, la sindrome di Tolosa Hunt si ripresenta con una ricaduta.
I sintomi neurologici sono causati dalla localizzazione locale del processo doloroso. Il dolore si manifesta a seguito dell'irritazione del primo ramo del nervo trigemino, che passa vicino al tronco del nervo oculomotore, e si manifesta a livello dell'orbita, della fronte, della tempia e della base del naso. L'intensità del dolore varia: da moderata a grave.
Sono possibili sintomi atipici, caratterizzati dall'assenza di dolore. Questo può essere osservato quando la lesione è localizzata prima dell'ingresso del quinto paio nel seno cavernoso.
I disturbi oculomotori si manifestano solitamente con una visione doppia durante la visione diretta.
Se il processo doloroso è localizzato nell'area dell'apice orbitario, le manifestazioni neurologiche si associano spesso a disturbi dell'analisi visiva. Di conseguenza, si verificano edema o atrofia del disco del nervo ottico e spesso si osserva uno scotoma centrale. Sono possibili esoftalmo (occhi sporgenti) e chemosi (edema congiuntivale), la cui insorgenza è causata da alterazioni infiltrative nel tessuto retrobulbare e da difficoltà di deflusso venoso dall'orbita.
Primi segni
Poiché la sindrome di Tolosa Hunt non è stata finora sufficientemente studiata, gli scienziati continuano a chiarire i possibili meccanismi di sviluppo di questa patologia. Tenendo conto dei criteri delineati dalla Società Neurologica Internazionale, la diagnosi di sindrome di Tolosa Hunt è giustificata dalla presenza di un granuloma della parete esterna del seno cavernoso, rilevato durante la risonanza magnetica cerebrale o una biopsia.
L'elenco dei segni accettati come criteri diagnostici per la sindrome è il seguente:
- dolore tipo "pizzico" o "torsione" in una cavità oculare con successivo sviluppo di paralisi muscolare (oftalmoplegia);
- lesioni combinate dei nervi oculomotori, del primo ramo del nervo trigemino e del plesso nervoso periarterioso;
- un peggioramento del quadro clinico nell'arco di diversi giorni (o entro 1-2 settimane);
- la possibilità di remissione spontanea (in alcuni casi – con conservazione residua dei difetti);
- la probabilità di una recidiva della sindrome, mesi o anni dopo;
- quadro sistemico immodificato, assenza di lesioni al di fuori del seno carotideo;
- la presenza di un effetto positivo della terapia corticosteroidea.
Esiste un altro elenco diagnostico simile di caratteristiche proposto nel 2003. Secondo questo elenco, la sindrome di Tolosa Hunt è considerata il risultato della proliferazione di tessuto granulomatoso nel seno cavernoso, nella fessura orbitaria superiore e nella cavità orbitaria:
- uno o più episodi di dolore monolaterale nella zona orbitale che si risolvono senza trattamento nel giro di un paio di settimane;
- danno al nervo cranico (III, IV o VI) sotto forma di paresi, presenza di un granuloma confermato da risonanza magnetica o biopsia;
- la comparsa della paresi contemporaneamente alla sindrome dolorosa o entro 14 giorni dalla stessa;
- scomparsa della paresi e della sindrome dolorosa entro 3 giorni dall'inizio della terapia corticosteroidea.
Forme
Nella sindrome di Tolosa Hunt, il lato sinistro e quello destro sono colpiti con una frequenza pressoché uguale, per cui la patologia viene distinta in sinistra o destra.
La malattia è solitamente monolaterale. Lesioni bilaterali sono state osservate solo in casi estremamente rari.
Il quadro clinico della malattia può svilupparsi attraverso le seguenti fasi:
- acuta o subacuta, che si verifica dopo una recente malattia infettiva virale, ipotermia, un forte aumento della pressione sanguigna, a volte senza una ragione evidente;
- cronica recidivante, con un graduale aumento dei sintomi e periodiche esacerbazioni.
Inoltre, la sindrome di Tolosa Hunt può essere:
- totale, con danno a tutti i nervi che passano attraverso la fessura orbitaria superiore;
- incompleto, con coinvolgimento nel processo patologico delle coppie di nervi VI, IV, III e del ramo I della coppia V in varie combinazioni.
Per quanto riguarda il seno, si possono distinguere le forme anteriore, media e posteriore della sindrome di Tolosa Hunt.
Complicazioni e conseguenze
La sindrome di Tolosa Hunt è accompagnata da forti dolori, che comportano perdita del sonno e disturbi della sfera emotiva e mentale. I malati diventano irritabili ed emotivamente instabili. Se non si interviene con il trattamento necessario, possono manifestarsi disturbi nevrotici: si sviluppano stati depressivi, nevrastenia e ipocondria. La capacità lavorativa si riduce significativamente e il paziente si isola.
Una caratteristica della sindrome di Tolosa Hunt è il suo decorso recidivante, tipico delle malattie autoimmuni. La durata del periodo di remissione può variare notevolmente: il massimo indicatore registrato di durata asintomatica è stato di 11 anni. Dopo il trattamento, il rischio di recidive si riduce significativamente. Le eventuali riacutizzazioni sono meno gravi.
Diagnostica Sindrome di Tolosa-Hunt
Spesso diventa difficile per i medici diagnosticare immediatamente la sindrome di Tolosa-Hunt, poiché i sintomi sono molto simili alle manifestazioni di altre malattie più comuni. Nella maggior parte dei casi, è necessario un ulteriore consulto con diversi specialisti: neurologo, oculista, endocrinologo, oncologo, neurochirurgo, ecc.
Nella prima fase è necessario escludere patologie maligne, aneurismi, meningiti, ecc.
Nella maggior parte dei casi, la sindrome di Tolosa Hunt viene diagnosticata per esclusione: il paziente si sottopone a una serie di esami per escludere altre patologie più probabili. Sono richiesti i seguenti esami:
- quadro ematico dettagliato;
- studio della funzione ormonale della tiroide;
- studio del livello delle proteine totali nel sangue (per valutare la qualità del metabolismo proteico);
- analisi del liquido cerebrospinale.
- La diagnostica strumentale prevede l'esecuzione delle seguenti procedure diagnostiche:
- risonanza magnetica dell'encefalo e della regione orbitaria, con e senza contrasto;
- angiografia a risonanza magnetica;
- angiografia sottrattiva digitale (angiografia sottrattiva endovenosa);
- Tomografia computerizzata dell'encefalo e dell'orbita con e senza contrasto.
La RM con gadolinio è la metodica di scelta per la valutazione della sindrome di Thalys e può dimostrare un ingrandimento e un enhancement anomali del CS che si estendono attraverso la fessura orbitaria superiore fino all'apice orbitario. I reperti RM riportati nelle immagini pesate in T1 e T2 sono estremamente variabili e aspecifici. La RM svolge un ruolo chiave nella diagnosi e aiuta a escludere altre lesioni comuni associate al CS, evitando la necessità di procedure invasive ad alto rischio come la biopsia del CS, l'unico modo per ottenere la conferma istopatologica di questa malattia.[ 5 ]
Questi studi aiutano a identificare tracce di processi infiammatori nel seno cavernoso, nella fessura orbitaria superiore o nell'apice orbitario. Tracce di infiammazione nella regione orbitaria su immagini trasversali in assenza di paralisi dei nervi cranici sono considerate più benigne in termini di prognosi.
Ad alcuni pazienti in cui si sospetta la sindrome di Tolosa Hunt viene consigliato di sottoporsi a una biopsia per escludere il cancro.
Diagnosi differenziale
La pratica clinica indica che sintomi simili possono essere presenti in molte patologie somatiche e neurologiche:
- nei processi infiammatori microbici, virali e fungini che colpiscono le meningi o la parete esterna del seno cavernoso;
- nei processi tumorali del cervello e dell'orbita - ad esempio, nell'adenoma pituitario, nel craniofaringioma, nel neurinoma, nel meningioma dell'ala dello sfenoide, nelle metastasi cerebrali o orbitali;
- nelle malformazioni vascolari, in particolare negli aneurismi venoso-arteriosi, nelle fistole carotido-cavernose, ecc., nonché nelle dissezioni dei rami dell'arteria carotide interna;
- per trombosi, formazioni cistiche del seno cavernoso, linfoma;
- per la sarcoidosi, la miosite orbitaria (muscoli oculari), la granulomatosi di Wegener (granulomatosi con poliangioite), l'oftalmoemicrania e alcune patologie del sangue.
La diagnosi differenziale consiste nell'elaborare la possibilità di sviluppare tutte le patologie sopra menzionate, sulla base dei risultati di un'indagine, di un esame obiettivo, di esami di laboratorio e strumentali.
Nella maggior parte dei casi, la sindrome di Tolosa Hunt deve essere distinta dalle seguenti patologie:
- ostruzione del seno cavernoso da parte di un trombo;
- Sindrome di Rochon-Duvignod;
- sindrome dello spazio retrosfenoidale (sindrome di Jacot);
- sindrome di Raeder paratrigeminale;
- polineuropatia cranica.
Chi contattare?
Trattamento Sindrome di Tolosa-Hunt
La sindrome di Tolosa Hunt risponde bene al trattamento con un ciclo di terapia immunosoppressiva a base di corticosteroidi. Questi farmaci sono in grado di sopprimere la risposta aggressiva del sistema immunitario e il suo effetto dannoso sui tessuti corporei.
I farmaci più comunemente prescritti sono Prednisolone, Metilprednisolone, Cortisone o farmaci alternativi che hanno mostrato effetti positivi nel trattamento di patologie autoimmuni note. I benefici degli steroidi sono probabilmente correlati al meccanismo antiossidante e/o alla capacità di dosi così elevate di ridurre l'edema e la successiva ischemia nelle aree colpite. [ 6 ]
Oltre ai corticosteroidi, è opportuno utilizzare antidolorifici e anticonvulsivanti. Sono obbligatori i preparati multivitaminici complessi.
Seguendo scrupolosamente tutte le istruzioni e le raccomandazioni del medico, i sintomi dolorosi della sindrome di Tolosa Hunt si attenuano rapidamente: i pazienti notano un netto miglioramento del loro benessere già dopo il secondo o terzo giorno. Nella stragrande maggioranza dei casi, la capacità lavorativa viene mantenuta. [ 7 ]
Dosaggi e frequenza ottimali di assunzione dei farmaci ormonali sono determinati su base individuale. Non esiste un regime terapeutico generalmente accettato, poiché è molto difficile organizzare studi controllati con placebo, il che è associato alla bassa prevalenza della sindrome. Il più delle volte, si raccomandano dosi elevate di corticosteroidi, sebbene vi siano stati casi di efficacia con dosi relativamente ridotte di farmaci (ad esempio, l'uso di Prednisolone in quantità inferiori a 0,5 mg/kg al giorno). Oggi, la quantità media di Prednisolone utilizzata nella sindrome di Tolosa-Hunt è di 1-2 mg/kg al giorno.
Piano di trattamento approssimativo:
- Metilprednisolone (Solu-Medron 1000 come infusione endovenosa con 250 ml di soluzione isotonica di cloruro di sodio e Panangin (10,0) al giorno per cinque giorni;
- Mildronate per la normalizzazione del metabolismo cellulare, 500 mg tramite iniezione endovenosa a getto al giorno per 10 giorni;
- Neuromidina per migliorare la trasmissione degli impulsi lungo le fibre neuromuscolari, 20 mg per via orale tre volte al giorno;
- Clonazepam per potenziare l'effetto inibitorio sulla trasmissione degli impulsi nervosi e sulla stimolazione dei recettori delle benzodiazepine, 2 mg per via orale, e/o Trileptal 150 mg per via orale prima di coricarsi.
È possibile prescrivere un ciclo prolungato di terapia glucocorticosteroidea utilizzando dosi elevate di Prednisolone. [ 8 ]
Prevenzione
Non è possibile prevenire in anticipo la sindrome di Tolosa Hunt. Questo è dovuto, almeno, al fatto che le cause del disturbo non sono ancora state chiaramente determinate. Se si riscontrano sintomi dolorosi, in particolare dolore frequente nella regione frontale e nelle orbite, visione doppia e indebolimento dei muscoli oculari, è necessario contattare lo specialista appropriato il prima possibile ed effettuare una diagnosi completa.
La prevenzione secondaria mira a prevenire le ricadute nei pazienti con sindrome di Tolosa-Hunt già diagnosticata. I punti importanti delle azioni preventive sono:
- consulenze mediche regolari, procedure diagnostiche e monitoraggio specialistico ambulatoriale;
- cicli periodici di terapia corticosteroidea;
- rafforzare e mantenere un adeguato stato del sistema immunitario.
Tutti i malati dovrebbero cercare di evitare situazioni stressanti e curare tempestivamente eventuali processi infiammatori nell'organismo.
Previsione
La prognosi della sindrome di Tolosa Hunt è considerata favorevole. La risposta alla terapia corticosteroidea è buona, i casi di remissione spontanea sono comuni, sebbene alcuni pazienti manifestino effetti residui sotto forma di compromissione della funzionalità dei muscoli oculari danneggiati. Se non trattata, la malattia diventa successivamente recidivante. Nei pazienti che hanno ricevuto il trattamento, si osservano recidive in circa il 35% dei casi. [ 9 ]
Dopo il completamento del percorso terapeutico, la capacità lavorativa viene solitamente ripristinata. Tuttavia, ciò vale per una malattia correttamente diagnosticata e non per altre patologie che si sviluppano sotto la “maschera” della sindrome. [ 10 ]
La disabilità si osserva solo in rari casi. Solo in caso di riacutizzazioni frequenti e documentate è possibile attribuire la disabilità al terzo gruppo. Nei casi più gravi, il paziente viene trasferito a lavori leggeri, non associati a carichi visivi. Se la sindrome di Tolosa Hunt è di natura persistente e ricorrente, al paziente è sconsigliata la guida di veicoli, a causa della compromissione della funzione motoria dei bulbi oculari e della diplopia.