Esperto medico dell'articolo

Nuove pubblicazioni
Sindrome di Angelman nei bambini e negli adulti
Ultima recensione: 04.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.

Esistono diverse malattie per le quali espressioni come "prenditi cura di te e non ti ammalerai" suonano quantomeno ridicole. Si tratta di patologie in cui alcune anomalie mentali e fisiche sono insite nel corpo del bambino già prima della nascita, ma i genitori non ne sono responsabili. Tali malattie sono causate da mutazioni o anomalie nei corredi cromosomici e sono chiamate cromosomiche o genetiche. Sindrome di Angelman, sindrome di Down, sindrome di Patau, sindrome di Edwards, sindrome di Turner, sindrome di Prader-Willi: queste sono solo alcune delle malattie genetiche di un elenco piuttosto nutrito.
Sindrome dell'uomo felice
Questa volta parleremo della patologia che prende il nome dal pediatra inglese Harry Angelman, che per primo sollevò la questione nel 1965, dopo aver incontrato il giorno prima nel suo studio tre bambini insoliti, accomunati da sintomi peculiari. Il medico chiamò questi bambini "bambini bambola" e scrisse un articolo su di loro, inizialmente intitolato "Bambini-marionette". L'articolo stesso e il suo titolo furono scritti ispirandosi a un dipinto visto in un museo di Verona. Il dipinto raffigurava un bambino che rideva, ed era intitolato "Il bambino burattino". L'associazione del bambino raffigurato nel dipinto con i tre bambini che Angelman aveva incontrato nel suo studio spinse il pediatra a raggrupparli in un unico gruppo a causa della patologia di cui erano affetti.
Non c'è nulla di sorprendente nel fatto che i bambini menzionati nell'articolo non siano stati notati da altri medici. Dopotutto, a prima vista sembrava che avessero malattie completamente diverse, tanto diverso era il quadro clinico generale della malattia in tre casi diversi. Forse la "nuova" patologia cromosomica avrebbe interessato altri scienziati, ma a quel tempo la genetica non era ancora sufficientemente sviluppata per confermare l'ipotesi del medico inglese. Pertanto, dopo un certo interesse, l'articolo fu a lungo accantonato.
La successiva menzione della sindrome di Angelman, come veniva ora chiamato l'articolo del pediatra inglese G. Angelman, risale ai primi anni '80 del XX secolo. E solo nel 1987 si è potuto scoprire il motivo per cui una piccola parte di bambini nasce con anomalie tali da sembrare, dall'esterno, sempre sorridenti e felici. In realtà, questo non è affatto vero, e il sorriso è solo una smorfia, dietro la quale si nasconde un'anima umana infelice e il dolore dei genitori.
Epidemiologia
Secondo le statistiche, una mutazione cromosomica in un bambino può svilupparsi sia in presenza di mutazioni simili nei genitori, sia in assenza di queste. Non esiste una chiara natura ereditaria della sindrome di Angelman (SA), ma la probabilità di sviluppare la patologia nei genitori con mutazioni cromosomiche è piuttosto elevata.
È anche interessante notare che se una famiglia ha già un figlio con SA, c'è una probabilità dell'1% di avere un secondo figlio con lo stesso disturbo, anche se i genitori sono sani.
Non esistono ancora statistiche precise sul numero di pazienti affetti dalla sindrome di Angelman. Forse il motivo risiede nella varietà dei sintomi, che possono presentarsi in una certa composizione o non presentarsi affatto per lungo tempo. Si stima che la prevalenza della malattia sia di 1 bambino ogni 20.000 neonati. Tuttavia, questa cifra è molto approssimativa.
Le cause Sindrome di Angelman
La sindrome di Angelman è il nome medico di una patologia cromosomica, ma non è l'unica. La si chiama sindrome dei bambini bambola, sindrome del pupazzo felice, sindrome di Petrushka e sindrome della bambola che ride. Si inventano nomi di ogni tipo (a volte persino offensivi per i pazienti stessi e i loro genitori), ma una malattia è una malattia, per quanto possa sembrare divertente e per quali ragioni.
E le cause dello sviluppo della sindrome di Angelman, come di molte altre patologie genetiche, sono in tutti i casi anomalie nella struttura di uno dei cromosomi o dell'intero corredo cromosomico. Ma nel nostro caso, il problema risiede interamente nel cromosoma 15, trasmesso dalla madre. In altre parole, il cromosoma paterno in questo caso non presenta deviazioni, mentre quello femminile subisce alcune mutazioni.
In base al tipo di anomalia cromosomica, la sindrome di Angelman è classificata come mutazione cromosomica. Tali mutazioni sono considerate:
- Una delezione (assenza di una sezione di un cromosoma contenente un certo insieme di geni; se uno dei geni manca, si parla di microdelezione), che è il risultato di due rotture e una riunione, quando una sezione del cromosoma originale viene persa.
- Duplicazione (presenza di una sezione extra in un cromosoma che è una copia di uno già esistente), che nella maggior parte dei casi porta alla morte della persona e, più raramente, all'infertilità.
- Inversione (inversione di una delle sezioni del cromosoma di 180 gradi, cioè nella direzione opposta, e quindi i geni in esso contenuti si trovano nell'ordine opposto), quando le estremità spezzate del cromosoma sono collegate in un ordine diverso dall'originale.
- Inserimento (se parte del materiale genetico in un cromosoma è fuori posto),
- traslocazione (se una certa sezione di un cromosoma è attaccata a un altro cromosoma; tale mutazione può essere reciproca senza perdita di sezioni).
Ricevendo un cromosoma mutato da una madre ignara, il bambino è destinato a nascere con delle anomalie. La causa più comune della sindrome di Angelman è ancora considerata la delezione del cromosoma 15 materno, ovvero la mancanza di una piccola sezione. Mutazioni meno comuni nella sindrome della "bambola che ride" sono considerate:
- traslocazione,
- disomia unipaterna (se il bambino ha ricevuto una coppia di cromosomi dal padre, il cromosoma materno è assente),
- mutazione dei geni nel DNA, che costituiscono sia il principale materiale costruttivo (genetico) sia le istruzioni per il suo corretto utilizzo (in particolare, mutazione del gene ube3a nel cromosoma materno).
La presenza di una di queste mutazioni nei genitori è un fattore di rischio per lo sviluppo della sindrome di Angelman nei bambini. Tuttavia, non solo le mutazioni cromosomiche, ma anche quelle genomiche (associate a un'alterazione quantitativa del corredo cromosomico e più comuni di quelle cromosomiche) possono provocare lo sviluppo della malattia in un bambino. Tra le mutazioni genomiche più comuni c'è la trisomia cromosomica (quando il corredo cromosomico di una persona ha più di 46 cromosomi).
Perché una patologia si manifesti in un bambino, non è affatto necessario che i genitori presentino anomalie cromosomiche. Eppure, esiste una certa percentuale di pazienti la cui malattia è ereditaria.
Patogenesi
Approfondiamo un po' la biologia, o più precisamente la genetica. L'informazione genetica di ogni singolo organismo umano è contenuta in 23 coppie di cromosomi. Un cromosoma di ogni coppia viene trasmesso al figlio dal padre, l'altro dalla madre. Tutte le coppie di cromosomi differiscono per forma e dimensioni e contengono determinate informazioni. Pertanto, la 23a coppia di cromosomi (cromosomi X e Y) è responsabile della formazione dei caratteri sessuali del bambino (XX - femmina, XY - maschio, mentre il cromosoma Y può essere ricevuto dal figlio solo dal padre).
Idealmente, un bambino riceve 46 cromosomi dai genitori, che determinano le sue caratteristiche genetiche, predeterminandolo come individuo. Un numero maggiore di cromosomi è chiamato trisomia ed è considerato una deviazione dalla norma. Ad esempio, la presenza del cromosoma 47 nel corredo cromosomico (cariotipo, che determina le caratteristiche di specie e individuali) causa l'insorgenza della sindrome di Down.
Colorando i cromosomi con un colorante speciale, al microscopio si possono osservare strisce di diverse tonalità lungo ciascuno di essi. All'interno di ogni striscia è presente un numero enorme di geni. Tutte queste strisce sono numerate dagli scienziati e hanno una posizione fissa. L'assenza di una delle strisce è considerata una deviazione dalla norma. Nella sindrome di Angelman, si può osservare molto spesso l'assenza di segmenti del cromosoma materno nell'intervallo q11-q13, situato nel braccio lungo, il cui numero di basi di DNA è di circa 4 milioni.
La componente principale del cromosoma è considerata una molecola di DNA incredibilmente lunga contenente migliaia di geni e decine e centinaia di milioni di basi azotate. Pertanto, il cromosoma 15, responsabile dello sviluppo della sindrome di Angelman e di molte altre, contiene 1200 geni e circa 100 milioni di basi. Qualsiasi alterazione della struttura della molecola di DNA influenzerà sicuramente l'aspetto e lo sviluppo del futuro bambino.
L'informazione genetica contenuta nei geni viene convertita in proteine o RNA. Questo processo è chiamato espressione genica. In questo modo, l'informazione genetica ricevuta dai genitori assume sia forma che contenuto, che si incarna nel loro erede unico, femminile o maschile.
Esistono numerose patologie con un tipo di ereditarietà non classico, tra cui la sindrome di Angelman, in cui i geni ricevuti dai genitori come parte di cromosomi appaiati portano un'impronta unica dei genitori e si manifestano in modi diversi.
La sindrome di Angelman è quindi un esempio lampante di imprinting genomico, in cui l'espressione genica nel corpo del bambino dipende direttamente dal genitore da cui sono stati ricevuti gli alleli (forme diverse di un gene, ricevute dal padre e dalla madre, localizzate su sezioni identiche di cromosomi appaiati). In altre parole, solo anomalie nel cromosoma materno portano allo sviluppo della sindrome, mentre mutazioni e anomalie strutturali del cromosoma paterno causano patologie completamente diverse.
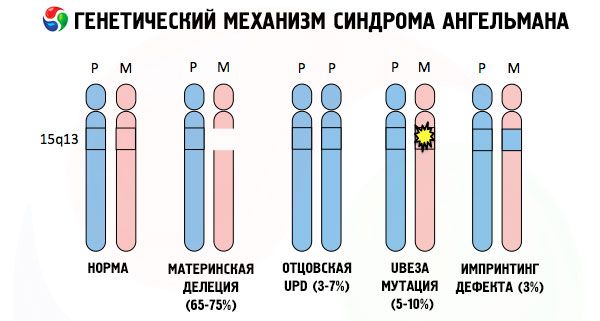
In questa patologia, si verifica la mancanza di alcuni geni nel cromosoma materno o una perdita/riduzione dell'attività di singoli geni (nella stragrande maggioranza dei casi, il gene ube3a, coinvolto nel metabolismo dell'ubiquitina, una proteina che regola la degradazione di altre proteine). Di conseguenza, al bambino vengono diagnosticate anomalie dello sviluppo mentale e deformità fisiche.
Sintomi Sindrome di Angelman
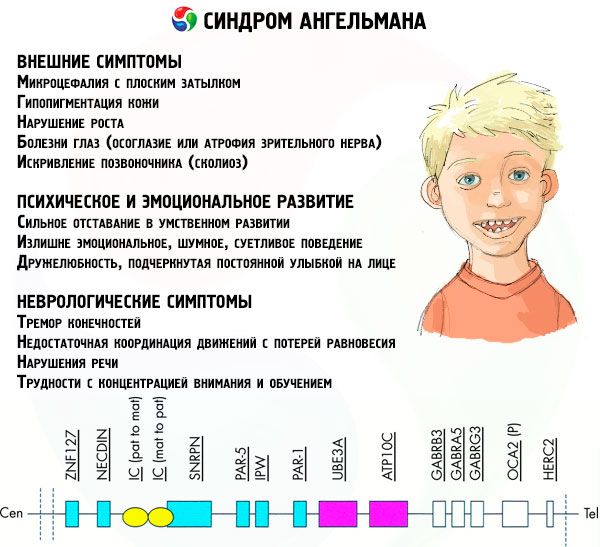
I sintomi della sindrome di Angelman influenzano vari aspetti della vita e dello sviluppo del bambino: fisico, neurologico e mentale. Sulla base di ciò, si possono identificare 3 gruppi di sintomi che indicano lo sviluppo di questa patologia.
- Sintomi esterni o fisici:
- una testa sproporzionatamente piccola rispetto al corpo e agli arti, che sono di dimensioni normali,
- bocca troppo larga,
- c'è quasi sempre un sorriso sul viso (con la bocca aperta),
- denti radi,
- labbro superiore stretto,
- lingua larga che sporge frequentemente,
- mascella inferiore sporgente,
- mento appuntito,
- pelle molto chiara, spesso con peli (albinismo, legato al fatto che il corpo non produce il pigmento melanina),
- macchie scure sulla pelle chiara (ipopigmentazione dovuta a insufficiente produzione di melanina)
- sintomi fisici o esterni: malattie oculari come strabismo o atrofia del nervo ottico,
- curvatura della colonna vertebrale (scoliosi),
- gambe rigide (quando cammina, la persona non piega le gambe all'altezza delle ginocchia a causa della scarsa mobilità delle articolazioni, da qui il paragone con l'andatura di una bambola).
- Sintomi correlati allo sviluppo mentale ed emotivo:
- ritardo mentale grave,
- comportamento eccessivamente emotivo, rumoroso, pignolo,
- frequenti battiti di mani,
- ha espresso cordialità, sottolineata da un sorriso costante sul viso,
- risate frequenti e senza motivo.
- Sintomi neurologici:
- tremore degli arti,
- insufficiente coordinazione dei movimenti con perdita di equilibrio,
- diminuzione del tono muscolare,
- vari disturbi del sonno,
- frequenti attacchi isterici nell'infanzia,
- disturbi del linguaggio (il bambino inizia a parlare tardi, ha scarse capacità comunicative e difficoltà di parola),
- iperattività sullo sfondo di aumentata eccitabilità,
- difficoltà di concentrazione e di apprendimento.
Ma questo è un quadro generalizzato della malattia. Infatti, il quadro clinico della sindrome di Angelman dipende in larga misura dallo stadio di sviluppo della malattia e dal tipo di mutazione cromosomica che l'ha causata. Ciò significa che i sintomi della malattia possono differire significativamente da paziente a paziente, il che per lungo tempo non ha permesso di distinguere la patologia da altre con un quadro clinico simile.
Tra i sintomi complessivi, possiamo evidenziare quelli che sono caratteristici di tutti i pazienti senza eccezioni:
- ritardo mentale grave,
- comportamento inappropriato (risate irragionevoli, aumento dell'eccitabilità, scarsa concentrazione, stato di euforia),
- sottosviluppo delle capacità motorie,
- scarsa coordinazione dei movimenti, andatura atassica (andatura irregolare, oscillazione da un lato all'altro, ecc.), tremore degli arti.
- disturbo dello sviluppo del linguaggio con predominanza di mezzi di comunicazione non verbali.
Tra i sintomi riscontrati dalla stragrande maggioranza dei pazienti si possono distinguere i seguenti:
- sproporzione tra la testa e il corpo causata da uno sviluppo fisico ritardato,
- in molti pazienti la forma del cranio è tale che le dimensioni del cervello rimangono più piccole rispetto alle persone sane (microcefalia),
- crisi epilettiche prima dei 3 anni di età con progressiva diminuzione di forza e frequenza in età avanzata,
- distorsione dei parametri EEG (fluttuazioni ed elevata ampiezza delle onde a bassa frequenza).
Questi sintomi sono piuttosto comuni, tuttavia il 20% dei pazienti affetti dalla sindrome di Angelman non ne è affetto.
Ancora meno frequentemente è possibile diagnosticare manifestazioni della malattia come:
- strabismo grave o lieve,
- scarso controllo del movimento della lingua, con conseguente frequente tirata fuori della lingua da parte dei pazienti senza motivo,
- difficoltà nella deglutizione e nella suzione, soprattutto nei bambini piccoli,
- alterazione della pigmentazione della pelle e degli occhi,
- braccia alzate o piegate mentre si cammina,
- iperreflessia,
- disturbi del sonno, soprattutto nell'infanzia,
- salivazione frequente,
- sete insaziabile,
- movimenti masticatori eccessivamente attivi,
- ipersensibilità al calore,
- parte posteriore piatta della testa,
- mascella inferiore sporgente,
- palmi lisci.
Una percentuale piuttosto elevata di pazienti presenta problemi di minzione, che non controllano correttamente, difficoltà motorie fini, che creano difficoltà nella cura di sé e nell'apprendimento, e sovrappeso. Quasi tutti i pazienti raggiungono la pubertà più tardi rispetto ai coetanei sani.
I bambini con sindrome di Angelman percepiscono bene il linguaggio orale e lo comprendono, ma non vogliono partecipare alle conversazioni, limitando il loro linguaggio a poche decine di parole, necessarie nella vita quotidiana. Tuttavia, in età adulta, questi pazienti sembrano più giovani rispetto ai loro coetanei senza patologie genetiche.
Molti sintomi della sindrome di Angelman sono incostanti, quindi il quadro clinico della malattia cambia significativamente con l'età. Convulsioni e crisi epilettiche diventano meno frequenti o scompaiono del tutto, il paziente diventa meno eccitabile e il sonno migliora.
Complicazioni e conseguenze
La sindrome di Angelman è una grave patologia cromosomica, attualmente praticamente incurabile, che priva i pazienti dell'opportunità di vivere una vita normale. La vita di un bambino con SA dipende in larga misura dal tipo di anomalia cromosomica.
La duplicazione di un segmento cromosomico è incompatibile con la vita nella maggior parte dei casi. E anche se questi pazienti non muoiono durante l'infanzia e raggiungono la pubertà, non hanno alcuna possibilità di avere figli.
La delezione o l'assenza di una parte dei geni che si verifica più frequentemente nella sindrome di Angelman rappresenta un ostacolo all'apprendimento del bambino a camminare e parlare. Questi bambini presentano una forma più grave di ritardo mentale e le crisi epilettiche si verificano più spesso e con un'intensità molto maggiore rispetto ai pazienti con altre anomalie cromosomiche.
Se la mutazione riguarda solo un gene, con la dovuta attenzione e il dovuto approccio è possibile insegnare al bambino le basi della cura di sé, della comunicazione e dell'interazione in gruppo, anche se rimarrà comunque indietro rispetto ai suoi coetanei nello sviluppo.
Per i bambini con sindrome di Angelman, che sono gentili per natura, la cosa più importante è l'amore e l'attenzione dei genitori. Solo in questo caso l'educazione del bambino darà i suoi frutti, anche se piccoli. Naturalmente, i pazienti con SA non potranno studiare in una scuola normale. Hanno bisogno di classi speciali in cui ai bambini verrà prima insegnato a concentrarsi e poi, gradualmente, verranno impartite le basi della conoscenza scolastica.
Diagnostica Sindrome di Angelman
La sindrome di Angelman è una patologia congenita dello sviluppo. Tuttavia, a causa di determinate circostanze, è spesso impossibile diagnosticarla durante l'infanzia e la prima infanzia. Ciò è dovuto alla non specificità e alla debole espressione dei sintomi nei neonati e nei bambini di età inferiore ai 3 anni. Inoltre, la prevalenza della malattia nel nostro Paese non è così elevata da permettere ai medici di riconoscerla tra i propri coetanei.
La sindrome di Angelman nei neonati può manifestarsi con una diminuzione del tono muscolare, che si manifesta con difficoltà di alimentazione (debolezza del riflesso di suzione e deglutizione) e, più tardi, difficoltà nell'apprendimento della deambulazione (questi bambini iniziano a camminare molto più tardi). Questi sintomi sono i primi segni di un'anomalia dello sviluppo nel neonato, che potrebbe essere associata a un'anomalia cromosomica. Solo l'analisi genetica può confermare questa ipotesi.
Particolare attenzione viene prestata ai bambini i cui genitori presentano diverse patologie genomiche o cromosomiche. Dopotutto, la malattia potrebbe non manifestarsi subito, ma se viene individuata in tempo, iniziando a lavorare intensamente con il bambino, è possibile ottenere un successo significativamente maggiore nell'apprendimento, rallentando la progressione della malattia.
Se i genitori presentano diverse anomalie cromosomiche, l'analisi genetica viene effettuata ancora prima della nascita del bambino, poiché l'SA è una delle patologie che possono essere rilevate nella fase embrionale.
La raccolta di materiale per la ricerca genetica può essere effettuata in due modi:
- invasiva (con una certa percentuale di rischio, poiché è necessario penetrare nell'utero per prelevare un campione di liquido amniotico),
- non invasivo (analisi del DNA del bambino dal sangue della madre).
Vengono poi effettuati i seguenti studi:
- ibridazione fluorescente in situ (metodo FISH): legame di una sonda di DNA marcata con un colorante speciale al DNA in studio, seguito da esame al microscopio.
- analisi delle mutazioni nel gene ube3a e nei geni imprinted,
- Analisi della metilazione del DNA mediante metodi speciali utilizzati in genetica.
I test genetici forniscono informazioni piuttosto accurate in caso di anomalie cromosomiche, il che significa che i futuri genitori sanno in anticipo a cosa prepararsi. Tuttavia, esistono delle eccezioni. In un certo gruppo di pazienti, in presenza di tutti i sintomi indicativi di patologia, i risultati dei test rimangono normali. In altre parole, la patologia può essere identificata solo osservando attentamente il bambino fin dalla prima infanzia: come mangia, quando ha iniziato a camminare e parlare, se piega le gambe quando cammina, ecc.
Oltre al metodo FISH, tra i metodi diagnostici strumentali per la sindrome di Angelman si possono distinguere la tomografia (TC o RM), che aiuta a determinare le condizioni e le dimensioni del cervello, e l'elettroencefalogramma (EEG), che mostra il funzionamento delle singole parti del cervello.
Di solito i medici formulano la diagnosi definitiva tra i 3 e i 7 anni, quando il paziente presenta già la maggior parte dei sintomi e sono visibili le dinamiche di sviluppo della malattia.
Quali test sono necessari?
Diagnosi differenziale
La sindrome di Angelman è una patologia genetica che non presenta praticamente manifestazioni specifiche. La maggior parte dei sintomi può essere indicativa sia della SA che di altre patologie genetiche.
La diagnosi differenziale della sindrome di Angelman viene effettuata con le seguenti patologie:
- Sindrome di Pitt-Hopkins (i pazienti sono caratterizzati da ritardo mentale, carattere allegro, sorriso, bocca piuttosto grande e larga, si nota microcefalia). La differenza sta negli attacchi di iperventilazione e apnea in stato di veglia.
- Sindrome di Christianson (i pazienti sono persone mentalmente ritardate, dal temperamento allegro, incapaci di parlare, caratterizzate da microcefalia, atassia, convulsioni, movimenti muscolari involontari).
- Sindrome di Mowat-Wilson (sintomi: ritardo mentale, crisi epilettiche, mento appuntito, bocca aperta, espressione felice, microcefalia). Segni particolari: grande distanza tra gli occhi, occhi obliqui verso l'interno, punta del naso arrotondata, padiglione auricolare rivolto all'indietro.
- Sindrome di Kabuki (caratterizzata da ritardo mentale da lieve a moderato, problemi motori e di linguaggio, debolezza muscolare, crisi epilettiche, microcefalia, lunghi intervalli tra i pruriti e difficoltà di coordinazione). È caratterizzata da sopracciglia arcuate, porzione laterale della palpebra inferiore estroflessa, occhi distanziati, lunghe fessure palpebrali con ciglia lunghe e folte.
- Sindrome di Rett (differenziata dalla sindrome di Asperger nelle donne). Sintomi: ritardo nello sviluppo del linguaggio, convulsioni, microcefalia. La differenza è che non c'è un'espressione felice sul viso, sono presenti attacchi di apnea e aprassia, che progrediscono nel tempo.
- Sindrome da ritardo mentale autosomica recessiva 38 (sintomi: ritardo mentale marcato con ritardi nelle capacità motorie e nel linguaggio, debolezza muscolare, problemi di alimentazione durante l'infanzia, impulsività). Caratteristica distintiva è il colore blu dell'iride.
- Sindrome da duplicazione genica MECP 2 (differenziazione da SA nei maschi). Sintomi: grave ritardo mentale, debolezza muscolare fin dall'infanzia, problemi di linguaggio o mancanza di linguaggio, epilessia. Distinzioni: miopatia progressiva, infezioni ricorrenti.
- Sindrome di Kleefstra (sintomi: difficoltà di linguaggio e pensiero, debolezza muscolare, disturbi del sonno, mancanza di attenzione, bocca aperta, iperattività, convulsioni, atassia, disturbi dell'equilibrio). Caratteristiche distintive: viso piatto, naso corto e camuso, occhi distanti, labbro inferiore grande e sporgente, scatti d'ira aggressivi.
- Sindrome di Smith-Magenis (caratterizzata da convulsioni, disturbi del sonno, disturbi dello sviluppo intellettivo e motorio). I tratti distintivi includono un viso largo e piatto e una fronte prominente.
- Sindrome di Koolen-de Vries (ritardo mentale da lieve a moderato, debolezza muscolare, convulsioni, socievolezza). Segni distintivi: viso allungato con fronte alta, orecchie sporgenti, occhi a mandorla, elevata mobilità articolare, difetti cardiaci congeniti.
- Sindrome di Phelan-McDermid (sintomi: ritardo mentale, disturbi del linguaggio o mancanza di linguaggio). Caratteristiche distintive: mani grandi con muscoli sviluppati, debolezza muscolare dalla nascita, sudorazione debole.
Patologie come la carenza di succinato di adenile, la sindrome da ritardo mentale autosomica recessiva di tipo 1, la sindrome da duplicazione del cromosoma 2q23.1, le sindromi da aploinsufficienza dei geni FOXG1, STXBP1 o MEF2C e alcune altre possono "vantare" sintomi simili alla sindrome di Angelman.
Il compito del medico è quello di formulare una diagnosi accurata, differenziando la sindrome di Angelman da patologie con sintomi simili e prescrivendo un trattamento efficace e adeguato allo stadio diagnosticato della malattia.
Trattamento Sindrome di Angelman
La sindrome di Angelman è una di quelle patologie per le quali la medicina è ancora alla ricerca di un trattamento efficace. Il trattamento eziologico della malattia è in fase di sviluppo con diversi metodi e mezzi, molti dei quali non sono ancora stati testati sull'uomo. Ciò significa che per ora i medici devono limitarsi alla terapia sintomatica, che contribuisce in qualche modo ad alleviare la situazione poco invidiabile di bambini e adulti affetti dalla sindrome della marionetta, che soffrono di crisi epilettiche, salivazione, ipotensione e disturbi del sonno.
Pertanto, è possibile ridurre la frequenza e l'intensità delle crisi epilettiche con l'ausilio di un farmaco anticonvulsivante opportunamente selezionato. Tuttavia, la difficoltà principale risiede nel fatto che le crisi nei pazienti con SA differiscono dalle crisi epilettiche ordinarie in quanto sono caratterizzate da diversi tipi di crisi, il che significa che la condizione può essere alleviata somministrando più farmaci contemporaneamente.
Gli anticonvulsivanti più comunemente utilizzati per il trattamento della sindrome di Angelman sono: acido valproico, topiramato, lamotrigina, levetiracetam, clonazepam e farmaci a base di questi. Meno comunemente utilizzati sono i farmaci a base di carmazepina, fenitoina, fenobarbital ed etosuccimide, poiché alcuni di essi possono provocare un effetto paradosso consistente nel potenziamento e nell'aumento della frequenza delle crisi epilettiche. Questo si verifica se il farmaco viene utilizzato in monoterapia.
Per trattare la scialorrea, si utilizzano solitamente due metodi: farmacologici (farmaci che sopprimono la produzione di saliva) e chirurgici, che prevedono il reimpianto dei dotti salivari. Tuttavia, nel caso della scialorrea, questi metodi sono considerati inefficaci e la questione rimane aperta. I genitori e coloro che si prendono cura di questi pazienti devono prestare particolare attenzione a questo problema, poiché i pazienti stessi di solito non riescono a controllare la scialorrea e alcuni non sono semplicemente in grado di prendersi cura di sé.
Un altro problema è la breve durata del sonno. Spesso i bambini con sindrome di Angelman non dormono più di 5 ore, il che ha un impatto negativo sul funzionamento di tutto l'organismo. I bambini facilmente eccitabili e attivi che amano giocare e comunicare (anche se cercano di limitarsi a metodi non verbali) sono visibilmente stanchi durante il giorno. Per riposare bene, il corpo ha bisogno di un sonno profondo e completo, ma è proprio questo il problema.
Sembrerebbe che i farmaci sedativi (fenotiazine e antipsicotici atipici) che calmano il sistema nervoso siano sufficienti a migliorare il sonno nei pazienti eccitabili. Tuttavia, nel caso della SA, l'uso di tali farmaci è gravato dal verificarsi di effetti negativi. Pertanto, i medici continuano a preferire sonniferi leggeri, come la melatonina (un farmaco ormonale naturale a base dell'ormone del sonno), che viene somministrata ai pazienti un'ora prima di coricarsi nella quantità di 1 compressa, e la difenidramina. La frequenza di somministrazione e il dosaggio sono stabiliti dal medico in base alle condizioni e all'età del paziente.
A volte i pazienti con sindrome di Angelman hanno problemi di digestione e feci. È possibile migliorare la digestione con lassativi (preferibilmente a base di erbe).
Oppure si può affrontare il problema in modo diverso, come hanno fatto i medici americani, basandosi su alcuni metodi di trattamento dell'autismo, poiché molti sintomi caratteristici della sindrome di Asperger sono tipici anche dell'autismo (impulsività, movimenti involontari, azioni ripetitive, deficit di attenzione, problemi di comunicazione, ecc.). È stato osservato che l'introduzione dell'ormone secretina, che normalizza la digestione e le feci, ha un effetto positivo sull'attenzione dei pazienti, e l'ossitocina contribuisce a migliorare le capacità cognitive e la memoria del bambino, nonché a correggere il comportamento.
È vero, gli ormoni da soli non bastano, soprattutto quando si tratta di bambini. Nella sindrome di Angelman, sono indicati la terapia comportamentale, il lavoro con uno psicologo e un logopedista (che insegni loro metodi di comunicazione non verbale e il linguaggio dei segni). L'educazione di questi bambini dovrebbe basarsi su un programma individuale con la partecipazione di insegnanti specializzati, uno psicologo e i genitori. Purtroppo, questo non è possibile ovunque e le famiglie vengono lasciate sole con il loro problema.
Poiché molti giovani pazienti con SA soffrono di basso tono muscolare e problemi articolari, si presta molta attenzione alla fisioterapia. Il più delle volte, i medici ricorrono ad applicazioni di paraffina, elettroforesi e magnetoterapia.
Il massaggio tonico attivo e specifici esercizi di fisioterapia aiuteranno il bambino malato a stare in piedi e a camminare con sicurezza dopo un po'. L'acquagym, particolarmente utile in questo senso, è consigliata per la sindrome di Asperger in acqua fredda. Aumenta il tono muscolare e insegna al bambino a controllare il proprio corpo e a coordinare i movimenti.
Trattamento anticonvulsivante
Il sintomo più pericoloso della sindrome di Angelman sono le crisi convulsive simili a quelle dell'epilessia. Questo sintomo si osserva nell'80% dei pazienti, il che significa che a tutti deve essere prescritto un trattamento anticonvulsivante efficace.
Il trattamento delle crisi epilettiche si effettua con l'ausilio di vitamine e anticonvulsivanti. Nella sindrome di Angelman, accompagnata da sindrome convulsiva, saranno utili le vitamine del gruppo B, così come le vitamine C, D ed E. Tuttavia, prescrivere autonomamente una terapia vitaminica in questo caso è molto pericoloso, poiché l'assunzione incontrollata di vitamine può ridurre l'efficacia dei farmaci antiepilettici e provocare nuove crisi, più gravi e prolungate.
Anche la scelta dei farmaci anticonvulsivanti e la prescrizione del loro dosaggio efficace dovrebbero essere effettuate da un medico specialista. Sarà lui a decidere se un solo farmaco sarà sufficiente o se il paziente dovrà assumerne due o più per un periodo prolungato.
Alla maggior parte dei pazienti, i medici prescrivono farmaci a base di acido valproico (Acido valproico, Depakine, Convulex, Valparin, ecc.), che prevengono le convulsioni e migliorano l'umore e lo stato mentale dei pazienti.
L'acido valproico è disponibile sotto forma di compresse, sciroppo e soluzioni iniettabili. Il farmaco più diffuso è il farmaco a rilascio prolungato "Depakine", disponibile in compresse e in soluzione per somministrazione endovenosa. Il dosaggio del farmaco viene determinato dal medico in base al peso, all'età e alle condizioni del paziente.
Il farmaco viene assunto durante i pasti, 2 o 3 volte al giorno. La dose giornaliera media è di 20-30 mg per 1 chilogrammo di peso corporeo del paziente, mentre la dose massima è di 50 mg/kg al giorno.
Controindicazioni all'uso. Non usare in caso di disfunzione epatica e pancreatica, diatesi emorragica, epatite, porfiria e ipersensibilità al farmaco.
Gli effetti collaterali includono tremori alle mani, disturbi digestivi e fecali e variazioni del peso corporeo.
Anche il topiramato è un farmaco di scelta per l'asma. È prodotto in compresse e viene utilizzato sia in monoterapia che in combinazione con altri farmaci.
Modo di somministrazione e dosaggio. Assumere le compresse per via orale indipendentemente dall'assunzione di cibo. La dose giornaliera iniziale per gli adulti è di 25-50 mg, per i bambini di 0,5-1 mg/kg. Il dosaggio viene aumentato settimanalmente secondo le indicazioni del medico.
Il farmaco non deve essere assunto durante la gravidanza e l'allattamento, così come in caso di ipersensibilità ai suoi componenti. Il farmaco ha molti effetti collaterali.
Farmaci che un medico può prescrivere per la sindrome di Angelman: Clomazepam, Rivotril, Lamotrigina, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra, ecc.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Medicina tradizionale e omeopatia
La medicina tradizionale, come i preparati omeopatici, è ovviamente relativamente sicura, ma l'efficacia di tale trattamento per la sindrome di Angelman può essere considerata controversa.
Sebbene la terapia popolare possa comunque essere d'aiuto in alcuni casi, stiamo parlando di fermare le crisi epilettiche. A questo proposito, la terapia a base di erbe può essere piuttosto efficace.
Un buon effetto è dato da una miscela di erbe medicinali a base di peonia, liquirizia e lenticchia d'acqua (i componenti vanno assunti in quantità uguali). Le erbe devono essere macinate fino a ottenere una farina. Dopo 2 settimane dall'inizio dell'assunzione, si può notare una significativa riduzione della frequenza delle crisi convulsive.
Anche il decotto di lavanda (1 cucchiaino per bicchiere di acqua bollente) è utile contro i crampi. La miscela viene fatta bollire per 5 minuti e lasciata in infusione per mezz'ora. Il rimedio va assunto la sera per 14 giorni.
L'infuso acquoso (o alcolico) di erba cardiaca è considerato efficace contro le crisi epilettiche.
Tra i preparati omeopatici per prevenire le convulsioni nella sindrome di Angelman, è possibile utilizzare medicinali a base di camomilla e cardiaca, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum e Arsenicum album. Tuttavia, è importante tenere presente che solo un medico omeopata può prescrivere dosi efficaci e sicure di farmaci in ogni caso specifico.
Prevenzione
Come probabilmente il lettore avrà già capito, la medicina non è ancora in grado di prevenire le mutazioni genetiche e altre anomalie cromosomiche, né di correggere la situazione. Questo può accadere a chiunque, perché i bambini con sindrome di Angelman nascono da genitori sani e la genetica, che è attualmente una delle branche della medicina meno studiate, non è ancora in grado di spiegare questo fenomeno.
L'unica cosa che si può fare è adottare un approccio responsabile alla pianificazione della gravidanza, registrarsi e sottoporsi agli esami per tempo. Ma, ancora una volta, tale misura sarà più educativa che preventiva, come qualsiasi esame. I giovani genitori sapranno in anticipo a cosa prepararsi e, in caso di risposta positiva, decideranno se possono assumersi la responsabilità di crescere un figlio malato.
Previsione
La prognosi della sindrome di Angelman dipende dalla natura dell'anomalia cromosomica e dalla tempestività della sua rilevazione. I più colpiti sono i bambini il cui cromosoma 15 presenta "lacune" genetiche (delezione). La probabilità che questi pazienti camminino e parlino è estremamente bassa. Altri casi possono essere corretti con un approccio attento e amore per il proprio figlio.
Purtroppo, questi pazienti non saranno in grado di diventare membri a pieno titolo della società, nonostante siano tutt'altro che stupidi e comprendano il linguaggio e il suo significato. Tuttavia, avranno problemi di comunicazione per il resto della loro vita. Ai pazienti può essere insegnato il linguaggio dei segni fin dall'infanzia, ma non possono essere costretti a comunicare usando le parole. Il vocabolario dei pazienti "parlanti" è limitato al minimo delle parole utilizzate nella vita quotidiana (5-15 parole).
Per quanto riguarda l'aspettativa di vita e lo stato di salute generale dei pazienti con sindrome di Angelman, i dati qui oscillano intorno ai valori medi. In età adulta, i pazienti affrontano principalmente problemi di salute come scoliosi e obesità, che, con il giusto approccio terapeutico, non sono pericolosi per la vita.