Esperto medico dell'articolo

Nuove pubblicazioni
Sindrome di Aper
Ultima recensione: 04.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.

 [
[ Le cause Sindrome di Aper
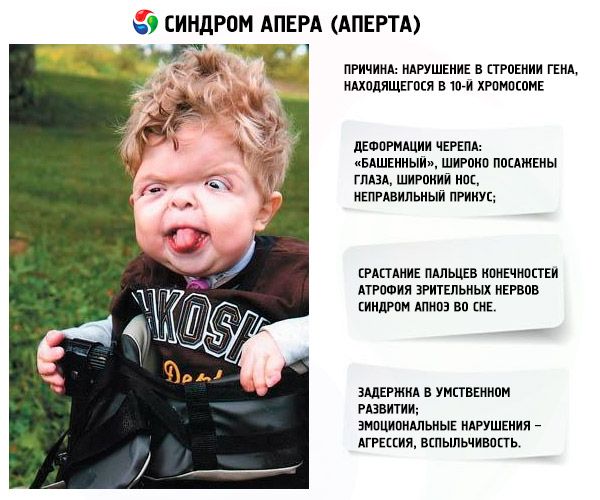
Lo sviluppo della sindrome è provocato da un disturbo nella struttura di un gene situato sul cromosoma 10. Questo gene è responsabile del processo di separazione delle dita e, inoltre, della chiusura tempestiva delle suture craniche.
Inoltre, le malattie infettive contratte durante la gravidanza (come rosolia o tubercolosi, sifilide o influenza, nonché meningite) e l'esposizione della madre ai raggi X sono considerate cause della patologia. Il più delle volte, tale sindrome viene riscontrata nei bambini nati da genitori anziani.
Patogenesi
La sindrome di Apert è ereditaria. La sua tipologia è autosomica dominante (vale a dire, in una famiglia in cui uno dei futuri genitori è malato, la probabilità di avere un figlio affetto da questa malattia è del 50-100%).
Una mutazione unica nel recettore 2 del fattore di crescita dei fibroblasti (FGFR2) determina un aumento del numero di cellule progenitrici che si sviluppano lungo il percorso osteogenico. Ciò si traduce in un aumento della formazione di matrice ossea sottoperiostea e in un'ossificazione prematura delle suture craniche durante lo sviluppo fetale. L'ordine e la velocità di fusione delle suture determinano il grado di deformità e disabilità. Una volta che il materiale di sutura è guarito, la crescita di altri tessuti perpendicolari a questa sutura è limitata e le ossa fuse agiscono come un'unica impalcatura ossea.
La prima prova genetica che la sindattilia nella sindrome di Apert è dovuta a un difetto del recettore del fattore di crescita dei cheratinociti (KGFR) è stata l'osservazione di una correlazione tra l'espressione di KGFR nei fibroblasti e la gravità della sindattilia.
L'ambliopia e lo strabismo sono più comuni nei pazienti con la mutazione Ser252Trp del gene FGFR2, mentre l'atrofia del disco ottico è più comune nei pazienti con la mutazione Pro253Arg del gene FGFR2. I pazienti con mutazioni Ser252Trp del gene FGR2 presentano una prevalenza significativamente maggiore di deficit visivo rispetto ai pazienti con la mutazione Pro253Arg del gene FGFR2.
Sintomi Sindrome di Aper
Alcune manifestazioni della malattia sono chiaramente evidenti già alla nascita del bambino, poiché si sviluppano all'interno dell'utero materno. Tra i principali sintomi della sindrome:
- Il cranio è deforme: si allunga in altezza, assumendo un aspetto "a torre"; inoltre, gli occhi sono distanziati e leggermente sporgenti (perché le dimensioni delle orbite diminuiscono); il naso si allarga e si forma una morso scorretta (i denti superiori sporgono eccessivamente);
- Le dita degli arti sono completamente fuse (principalmente l'anulare, ma anche il medio e l'indice) e sembrano una membrana cutanea o una fusione completa di ossa; inoltre, possono crescere dita extra;
- Ritardo mentale (non si verifica in tutti);
- Atrofia dei nervi ottici, con conseguente diminuzione dell'acuità visiva (in alcuni casi si arriva alla perdita completa della vista);
- L'aumento della pressione intracranica, che si verifica a seguito della chiusura prematura delle suture craniche, si manifesta sotto forma di mal di testa e vomito con nausea;
- Poiché la mascella superiore rimane sottosviluppata, sorgono problemi respiratori;
- La sindrome dell'apnea notturna è un fenomeno comune.
- Manifestazioni emotive: aggressività, mancanza di controllo, forte irascibilità.
Complicazioni e conseguenze
La maggior parte dei pazienti presenta un certo grado di ostruzione delle vie aeree superiori durante l'infanzia. Le vie aeree superiori sono scarsamente pervie a causa delle dimensioni ridotte del rinofaringe e delle coane, mentre le vie aeree inferiori possono causare morte precoce a causa di anomalie della cartilagine tracheale.
Diagnostica Sindrome di Aper
Per effettuare una diagnosi sono necessari i seguenti passaggi:
- Il medico dovrebbe analizzare i disturbi lamentati dal paziente, così come la sua storia clinica. È necessario scoprire se in famiglia si sono verificati casi di patologie simili;
- Esame neurologico per valutare la forma del cranio e lo sviluppo intellettivo del paziente (questionari specifici e colloquio);
- Esame del fondo dell'occhio per rilevare la presenza di sintomi di aumento della pressione intracranica (gonfiore della papilla ottica e offuscamento dei suoi bordi);
- Per valutare le condizioni del cranio viene eseguita una radiografia;
- Risonanza magnetica e computerizzata della testa per esaminare la struttura del cervello e del cranio strato per strato, per determinare la presenza di sintomi di fusione prematura delle suture craniche e, inoltre, idrocefalo (a causa dell'aumento della pressione intracranica, si accumula un eccesso di liquido cerebrospinale (si tratta del liquido cerebrospinale che favorisce il processo metabolico e la nutrizione del cervello));
- Radiografia dei piedi e delle mani per determinare la causa della fusione delle dita (importante per pianificare il successivo intervento chirurgico);
- Potrebbe essere prescritta una consulenza con un genetista medico e un neurochirurgo.
Test
È in corso l'analisi genetica delle mutazioni comuni che si verificano nel gene di tipo FGFR2.
Diagnosi differenziale
Questa sindrome deve essere differenziata da altre patologie genetiche in cui si osserva craniosinostosi, come le sindromi di Pfeiffer, Crouzon, Saethre-Chotzen e Carpenter. Per escludere queste anomalie, si utilizzano metodi di genetica molecolare.
Chi contattare?
Trattamento Sindrome di Aper
L'intervento chirurgico è considerato l'unico trattamento efficace per la sindrome di Apert: aiuta a correggere i difetti fisici individuali e corregge anche il ritardo mentale.
Durante questa procedura, la sutura coronale viene chiusa per prevenire possibili lesioni cerebrali. La tecnica più comune è la distrazione craniofacciale, che prevede una trazione graduale del cranio. Per rimuovere i singoli difetti facciali, si esegue un intervento di chirurgia ortodontica e/o ortognatica.
Inoltre, i pazienti vengono sottoposti a correzione chirurgica della fusione delle dita.
Riferimenti
Genetica medica. Guida nazionale. A cura di Ginter EK, Puzyrev VP GEOTAR-Media. 2022