Esperto medico dell'articolo

Nuove pubblicazioni
Polimiosite e dermatomiosite: cause, sintomi, diagnosi, trattamento
Ultima recensione: 05.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
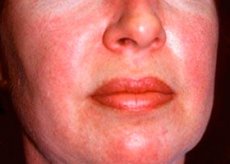
Polimiosite e dermatomiosite sono rare malattie reumatiche sistemiche caratterizzate da alterazioni infiammatorie e degenerative a carico dei muscoli (polimiosite) o di muscoli e pelle (dermatomiosite). La manifestazione cutanea più specifica è l'eruzione cutanea eliotropa.
Il coinvolgimento muscolare è simmetrico e comprende debolezza, dolorabilità e successiva atrofia dei muscoli prossimali del cingolo pelvico. Le complicazioni possono includere il coinvolgimento viscerale e la malignità. La diagnosi si basa sulla presentazione clinica e sulla valutazione della disfunzione muscolare mediante misurazione dei livelli enzimatici, esecuzione di risonanza magnetica, elettromiografia e biopsia muscolare. Il trattamento prevede glucocorticoidi, talvolta in combinazione con immunosoppressori o immunoglobuline per via endovenosa.
Le donne si ammalano due volte più spesso degli uomini. La malattia può manifestarsi a qualsiasi età, ma viene diagnosticata più frequentemente tra i 40 e i 60 anni; nei bambini, tra i 5 e i 15 anni.
 [
[ Quali sono le cause della dermatomiosite e della polimiosite?
Si ritiene che la causa della malattia sia una reazione autoimmune al tessuto muscolare in individui geneticamente predisposti. La malattia è più comune in presenza di una storia familiare negativa e di portatori di determinati antigeni HLA (DR3, DR52, DR56). Possibili fattori scatenanti sono la miosite virale e le neoplasie maligne. Sono stati segnalati casi di rilevamento di strutture simili ai picornavirus nelle cellule muscolari; inoltre, i virus possono indurre malattie simili negli animali. L'associazione di tumori maligni con la dermatomiosite (molto meno frequente rispetto alla polimiosite) suggerisce che la crescita tumorale possa anche essere un fattore scatenante per lo sviluppo della malattia, a seguito dell'avvio di reazioni autoimmuni ad antigeni comuni del tumore e del tessuto muscolare.
Depositi di IgM, IgG e della terza componente del complemento si trovano nelle pareti dei vasi sanguigni dei muscoli scheletrici; questo è particolarmente caratteristico della dermatomiosite nei bambini. I pazienti con polimiosite possono anche sviluppare altri processi autoimmuni.
Fisiopatologia della dermatomiosite e della polimiosite
Le alterazioni patologiche includono danni cellulari e atrofia su uno sfondo di infiammazione di diversa gravità. I muscoli degli arti superiori e inferiori, così come quelli del viso, sono danneggiati in misura minore rispetto ad altri muscoli scheletrici. Le lesioni ai muscoli viscerali della faringe e dell'esofago superiore, e meno frequentemente a quelli di cuore, stomaco o intestino, possono portare a disfunzioni degli organi sopra menzionati. Elevate concentrazioni di mioglobina dovute a rabdomiolisi possono causare danni renali. Possono verificarsi anche alterazioni infiammatorie a carico di articolazioni e polmoni, soprattutto nei pazienti con anticorpi antisintetasi.
Sintomi di dermatomiosite e polimiosite
L'esordio della polimiosite può essere acuto (soprattutto nei bambini) o subacuto (più spesso negli adulti). Un'infezione virale acuta talvolta precede o è un fattore scatenante per la manifestazione della malattia, le cui manifestazioni più comuni sono debolezza dei muscoli prossimali o eruzioni cutanee. Il dolore si manifesta in misura minore rispetto alla debolezza. Possono svilupparsi poliartralgia, fenomeno di Raynaud, disfagia, disturbi polmonari e sintomi generali (aumento della temperatura corporea, calo del peso corporeo, debolezza). Il fenomeno di Raynaud si riscontra spesso in pazienti con concomitanti malattie del tessuto connettivo.
La debolezza muscolare può progredire nell'arco di diverse settimane o mesi. Tuttavia, affinché si manifesti clinicamente, è necessario che sia interessato almeno il 50% delle fibre muscolari (pertanto, la presenza di debolezza muscolare indica la progressione della miosite). I pazienti possono avere difficoltà a sollevare le braccia al di sopra del livello delle spalle, a salire le scale o ad alzarsi da una posizione seduta. A causa della grave debolezza dei muscoli pelvici e del cingolo scapolare, i pazienti possono essere costretti a una sedia a rotelle o a letto. Se i flessori del collo sono interessati, diventa impossibile sollevare la testa dal cuscino. L'interessamento dei muscoli della faringe e dell'esofago superiore porta a disturbi della deglutizione e rigurgito. I muscoli degli arti inferiori, superiori e del viso solitamente non sono interessati. Tuttavia, possono svilupparsi contratture degli arti.
Le eruzioni cutanee osservate nella dermatomiosite sono solitamente di colore scuro ed eritematose. È caratteristico anche l'edema periorbitale di colore violaceo (eritema eliotropo). Le eruzioni cutanee possono essere leggermente sollevate rispetto al livello cutaneo ed essere lisce o squamose; le localizzazioni delle eruzioni cutanee sono fronte, collo, spalle, torace, schiena, avambracci, parte inferiore degli stinchi, sopracciglia, area del ginocchio, malleoli mediali, superfici dorsali delle articolazioni interfalangee e metacarpofalangee, sul lato laterale (sintomo di Gottron). È possibile iperemia della base o della periferia delle unghie. Una dermatite desquamativa, accompagnata da fissurazioni, può svilupparsi sulla cute della superficie laterale delle dita. Le lesioni cutanee primarie spesso si risolvono senza sequele, ma possono portare ad alterazioni secondarie come pigmentazione scura, atrofia, cicatrici o vitiligine. Possono svilupparsi calcificazioni sottocutanee, soprattutto nei bambini.
Circa il 30% dei pazienti sviluppa poliartralgia o poliartrite, spesso accompagnata da edema e versamento articolare. Tuttavia, la gravità delle manifestazioni articolari è modesta. Si verificano più frequentemente quando nei pazienti vengono rilevati anticorpi anti-Jo-1 o altre sintetasi.
Il coinvolgimento degli organi interni (ad eccezione della faringe e dell'esofago superiore) è meno comune nella polimiosite rispetto ad altre malattie reumatiche (in particolare LES e sclerosi sistemica). Raramente, soprattutto nella sindrome da antisintetasi, la malattia si manifesta come polmonite interstiziale (sotto forma di dispnea e tosse). Possono svilupparsi aritmie cardiache e disturbi della conduzione, ma sono generalmente asintomatici. Le manifestazioni gastrointestinali sono più comuni nei bambini affetti anche da vasculite e possono includere vomito con sangue, melena e perforazione intestinale.
Dove ti fa male?
Classificazione della polimiosite
Esistono 5 tipi di polimiosite.
- La polimiosite idiopatica primaria, che può manifestarsi a qualsiasi età, non coinvolge la pelle.
- La dermatomiosite idiopatica primaria è simile alla polimiosite idiopatica primaria, ma interessa la pelle.
- Polimiosite e dermatomiosite associate a neoplasie maligne possono verificarsi in pazienti di qualsiasi età; il loro sviluppo è più spesso osservato nei pazienti anziani, così come in pazienti con altre malattie del tessuto connettivo. Lo sviluppo di neoplasie maligne può essere osservato sia entro 2 anni prima che entro 2 anni dopo l'insorgenza della miosite.
- La polimiosite o dermatomiosite infantile è associata alla vasculite sistemica.
- La polimiosite e la dermatomiosite possono manifestarsi anche in pazienti affetti da altre malattie del tessuto connettivo, le più comuni delle quali sono la sclerosi sistemica progressiva, la malattia mista del tessuto connettivo e il lupus eritematoso sistemico (LES).
L'inclusione della miosite dei muscoli del tronco nel gruppo delle polimiositi è errata, poiché quest'ultima è una malattia distinta caratterizzata da manifestazioni cliniche simili a quelle della polimiosite idiopatica cronica. Tuttavia, si sviluppa in età avanzata, colpisce spesso i muscoli delle parti distali del corpo (ad esempio, gli arti superiori e inferiori), ha una durata maggiore, risponde meno bene al trattamento ed è caratterizzata da un quadro istologico tipico.
Diagnosi di dermatomiosite e polimiosite
La polimiosite deve essere sospettata nei pazienti che lamentano debolezza muscolare prossimale, con o senza dolorabilità. La valutazione per la dermatomiosite è necessaria nei pazienti che lamentano un'eruzione cutanea simile all'eliotropio o al segno di Gottron, così come nei pazienti con manifestazioni di polimiosite associate a lesioni cutanee compatibili con la dermatomiosite. Le manifestazioni cliniche di polimiosite e dermatomiosite possono assomigliare a quelle della sclerosi sistemica o, meno comunemente, del lupus eritematoso sistemico (LES) o della vasculite. La certezza della diagnosi aumenta soddisfacendo il maggior numero possibile dei seguenti cinque criteri:
- debolezza dei muscoli prossimali;
- eruzioni cutanee caratteristiche;
- aumento dell'attività degli enzimi del tessuto muscolare (creatina chinasi o, in assenza di aumento della sua attività, aminotransferasi o aldolasi);
- cambiamenti caratteristici nella miografia o nella risonanza magnetica;
- cambiamenti istologici caratteristici in una biopsia del tessuto muscolare (criterio assoluto).
La biopsia muscolare può escludere alcune condizioni clinicamente simili, come la miosite dei muscoli del tronco e la rabdomiolisi dovuta a infezione virale. Le alterazioni rilevate dall'esame istologico possono variare, ma l'infiammazione cronica e i focolai di degenerazione e rigenerazione muscolare sono tipici. Una diagnosi accurata (solitamente mediante verifica istologica) è necessaria prima di iniziare un trattamento potenzialmente tossico. La risonanza magnetica (RM) può rilevare focolai di edema e infiammazione nei muscoli, seguiti da una biopsia mirata.
Gli esami di laboratorio possono confermare o, al contrario, escludere il sospetto di presenza della malattia e sono utili anche per valutarne la gravità, la possibilità di una combinazione con altre patologie simili e per diagnosticare le complicanze. Nonostante in alcuni pazienti vengano rilevati anticorpi antinucleari, questo fenomeno è più tipico di altre malattie del tessuto connettivo. Circa il 60% dei pazienti presenta anticorpi contro l'antigene dei nuclei (PM-1) o delle cellule del timo intero e Jo-1. Il ruolo degli autoanticorpi nella patogenesi della malattia rimane poco chiaro, sebbene sia noto che gli anticorpi contro Jo-1 sono un marcatore specifico della sindrome da antisintetasi, che comprende alveolite fibrosante, fibrosi polmonare, artrite e fenomeno di Raynaud.
La valutazione periodica dell'attività della creatinchinasi è utile per il monitoraggio del trattamento. Tuttavia, nei pazienti con grave atrofia muscolare, l'attività enzimatica può essere normale nonostante la presenza di miosite cronica attiva. La risonanza magnetica, la biopsia muscolare o un'elevata attività della creatinchinasi sono spesso utili per distinguere tra una recidiva di polimiosite e una miopatia indotta da glucocorticoidi.
Poiché molti pazienti presentano neoplasie maligne non diagnosticate, alcuni autori raccomandano di sottoporre a screening tutti gli adulti con dermatomiosite e quelli con polimiosite di età superiore ai 60 anni, seguendo il seguente schema: esame obiettivo, inclusi esami mammari, pelvici e rettali (incluso il test delle feci per la ricerca del sangue occulto); emocromo completo; emochimica; mammografia; test dell'antigene carcinoembrionario; analisi delle urine; radiografia del torace. Alcuni autori mettono in dubbio la necessità di tale screening nei pazienti più giovani che non presentano evidenze cliniche di neoplasia maligna.
Cosa c'è da esaminare?
Trattamento della dermatomiosite e della polimiosite
Finché l'infiammazione non si è attenuata, l'attività fisica deve essere limitata. I glucocorticoidi sono farmaci di prima linea. Nella fase acuta della malattia, ai pazienti adulti deve essere prescritto prednisolone (per via orale) a una dose da 40 a 60 mg al giorno. La determinazione regolare dell'attività della creatinchinasi è un indicatore precoce di efficacia: nella maggior parte dei pazienti, si nota una diminuzione o una normalizzazione entro 6-12 settimane a seguito di un aumento della forza muscolare. Dopo la normalizzazione dell'attività enzimatica, la dose di prednisolone viene ridotta: inizialmente di circa 2,5 mg al giorno per una settimana, poi più rapidamente; se l'attività enzimatica muscolare aumentata si ripresenta, la dose ormonale viene nuovamente aumentata. I pazienti guariti possono fare a meno dei glucocorticoidi, ma il più delle volte i pazienti adulti necessitano di una terapia glucocorticoide a lungo termine (10-15 mg di prednisolone al giorno). La dose iniziale di prednisolone per i bambini è di 30-60 mg/m² una volta al giorno. In presenza di remissione per > 1 anno nei bambini, la terapia con glucocorticoidi può essere interrotta.
In alcuni casi, i pazienti che ricevono dosi elevate di glucocorticoidi manifestano un improvviso aumento della debolezza muscolare, che può essere associato allo sviluppo di miopatia da glucocorticoidi.
In caso di risposta inadeguata al trattamento con glucocorticoidi, così come in caso di sviluppo di miopatia da glucocorticoidi o altre complicazioni che richiedono una riduzione del dosaggio o l'interruzione del prednisolone, si devono utilizzare immunosoppressori (metotrexato, ciclofosfamide, azatioprina, ciclosporina). Alcuni pazienti possono ricevere solo metotrexato (solitamente a dosi superiori a quelle utilizzate per il trattamento dell'artrite reumatoide) per più di 5 anni. Le immunoglobuline per via endovenosa possono essere efficaci nei pazienti refrattari alla terapia farmacologica, ma il loro utilizzo aumenta il costo del trattamento.
La miosite associata a tumori primari e metastatici, così come la miosite dei muscoli del tronco, sono solitamente più refrattarie alla terapia con glucocorticoidi. La remissione della miosite associata a tumori maligni è possibile dopo l'asportazione del tumore.
Qual è la prognosi per la dermatomiosite e la polimiosite?
Una remissione a lungo termine (e persino una guarigione clinica) nell'arco di 5 anni si osserva in oltre la metà dei pazienti trattati; nei bambini, questa percentuale è più alta. La recidiva, tuttavia, può verificarsi in qualsiasi momento. Il tasso di sopravvivenza complessivo a cinque anni è del 75%, più alto nei bambini. Le cause di morte negli adulti sono debolezza muscolare grave e progressiva, disfagia, deficit nutrizionale, polmonite ab ingestis o insufficienza respiratoria dovuta a infezioni polmonari. La polimiosite è più grave e resistente al trattamento in presenza di danni a cuore e polmoni. Nei bambini, la morte può verificarsi a causa di vasculite intestinale. La prognosi generale della malattia è determinata anche dalla presenza di neoplasie maligne.