Esperto medico dell'articolo

Nuove pubblicazioni
L'alcaptonuria è un'anomalia enzimatica congenita
Ultima recensione: 04.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
Uno dei disturbi metabolici più rari, l'alcaptonuria, è un'anomalia congenita nel metabolismo dell'amminoacido tirosina.
Questa sindrome può anche essere chiamata deficit di omogentisato ossidasi, omogentisinuria, ocronosi ereditaria o malattia delle urine nere.[ 1 ]
Epidemiologia
Secondo le statistiche, non si registrano più di nove casi di alcaptonuria ogni milione di persone. E nella maggior parte dei paesi europei, si registra un caso ogni 100-250.000 nati vivi.
Tra i paesi europei, l'eccezione è la Slovacchia (in particolare la relativamente piccola regione nord-occidentale), dove la prevalenza di alcaptonuria è di un caso ogni 19.000 neonati. Ciò è molto probabilmente dovuto al fatto che tra le famiglie Rom slovacche che vi risiedono, il livello di consanguineità (matrimoni tra cugini) è il più alto in Europa: 10-14%. [ 2 ]
Le cause alcaptonuria
Le cause esatte dell'alcaptonuria, come disturbo congenito del catabolismo (scomposizione metabolica) dell'α-amminoacido aromatico (omociclico) tirosina, sono state stabilite: questo tipo di disturbo metabolico è una conseguenza di mutazioni omozigoti o eterozigoti composte di uno delle migliaia di geni sul cromosoma 3, più precisamente, del gene HGD nel locus 3q21-q23 sul braccio lungo del cromosoma. Questo gene codifica le sequenze nucleotidiche dell'enzima epatico omogentisato-1,2-diossigenasi [ 3 ] (chiamato anche ossidasi dell'acido omogentisico o omogentisato ossidasi) - una metalloproteina contenente ferro necessaria per una delle fasi della degradazione della tirosina nel corpo. [ 4 ], [ 5 ]
L'alcaptonuria è quindi un difetto dell'enzima omogentisato-1,2-diossigenasi, o più precisamente, il risultato della sua carenza geneticamente determinata o della sua completa assenza. [ 6 ]
Essendo una carenza enzimatica congenita, l'alcaptonuria viene ereditata come tratto autosomico recessivo, ovvero affinché l'alcaptonuria si manifesti nei bambini, entrambi i genitori devono avere un gene modificato per l'enzima, poiché ciascuno di essi trasmette al figlio solo una copia del gene delle due disponibili.
Secondo i dati più recenti, esistono più di duecento varianti di modificazione del gene HGD e le mutazioni missense, la traslocazione e lo splicing sono quelle osservate più frequentemente.
Fattori di rischio
L'unico fattore di rischio per lo sviluppo di questa enzimopatia congenita è la sua presenza nella storia familiare e l'ereditarietà di due copie modificate del gene HGD, se i genitori non presentano alcaptonuria (il rischio di trasmettere l'anomalia è del 25%), o uno dei genitori è affetto da questo disturbo. [ 7 ]
Patogenesi
La tirosina svolge un ruolo fondamentale nella sintesi delle proteine, nella produzione delle cromoproteine (la melanina, il pigmento della pelle), nonché degli ormoni tiroidei e dei neurotrasmettitori catecolamine.
Il meccanismo di regolazione della quantità di tirosina nelle cellule è molto complesso e l'organismo ne normalizza l'eccesso scomponendola. Il processo di catabolismo della tirosina, come quello di tutti gli amminoacidi aromatici, è multistadio e si svolge in diverse fasi. Ogni fase della degradazione metabolica della tirosina avviene con l'intervento di uno specifico enzima e la formazione di un composto intermedio.
Quindi, prima l'amminoacido viene scomposto in para-idrossifenilpiruvato, che viene convertito in alcaptone – acido 2,5-diidrossifenilacetico o omogentisico. Poi l'alcaptone dovrebbe essere trasformato in acido maleacetico, ma ciò non accade. [ 8 ]
La patogenesi dell'alcaptonuria consiste nell'interruzione delle reazioni biochimiche del catabolismo della tirosina nella fase di formazione dell'acido omogentisinico: semplicemente non è necessario alcun enzima per scomporlo: l'omogentisato ossidasi.
L'acido omogentisico non viene utilizzato dall'organismo e può accumularsi con l'escrezione renale. Inoltre, viene ossidato a benzochinoacetato (acido benzochinoneacetico) che, legandosi alle molecole dei tessuti e dei fluidi corporei, forma composti biopolimerici colorati come la melanina.
L'accumulo di questi prodotti intermedi nel tessuto porta alla rottura della struttura del collagene del tessuto cartilagineo, con conseguente riduzione della sua elasticità, con la comparsa di numerosi segni clinici di alcaptonuria e lo sviluppo di complicazioni.
Sintomi alcaptonuria
L'alcaptonuria nei neonati e nei bambini è caratterizzata dall'imbrunimento delle urine. Quando esposte all'aria, le urine su pannolini, pannolini e biancheria intima diventano marrone scuro; ciò è dovuto all'accumulo e al rilascio di acido omogentisinico, che viene ossidato a benzochinoacetato. [ 9 ]
In assenza di altri sintomi, l'alcaptonuria nei bambini piccoli spesso non viene riconosciuta tempestivamente, poiché l'urina può scurirsi dopo diverse ore dalla minzione. Secondo alcuni dati, solo un quinto dei bambini di età inferiore ai 12 mesi nati con questo deficit enzimatico viene identificato in ambito clinico. Pertanto, è molto importante che i genitori prestino attenzione alla cura dei loro neonati.
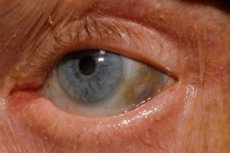
Inoltre, i primi segni includono la pigmentazione (colore grigio-bluastro) della sclera degli occhi e delle cartilagini delle orecchie e del naso, che spesso viene chiamata ocronosi.[ 10 ]
Con il passare del tempo compaiono altri sintomi:
- grave pigmentazione della pelle sugli zigomi, sulle ascelle e sui genitali;
- macchie sugli indumenti a contatto con le zone sudate del corpo;
- attacchi di debolezza generale;
- voce roca.
Occorre tenere presente che alcaptonuria e ocronosi, come sopra menzionato, sono nomi sinonimi per lo stesso disturbo del catabolismo della tirosina.
Malattia delle urine a sciroppo d'acero e alcaptonuria. La malattia congenita delle urine a sciroppo d'acero, o leucinosi, è anch'essa un disturbo metabolico, ha lo stesso modello di ereditarietà e persino le mutazioni si verificano sullo stesso cromosoma, ma colpiscono il gene che codifica per il complesso enzimatico dell'α-chetoacido deidrogenasi a catena ramificata. Per questo motivo, l'organismo non riesce a scomporre alcuni componenti delle proteine, in particolare gli amminoacidi leucina, isoleucina e valina. In questa malattia, l'urina (e il cerume) hanno un odore dolce; inoltre, il quadro clinico di questo tipo di acidemia organica include ipopigmentazione, fluttuazioni della pressione sanguigna, convulsioni, vomito e diarrea, calo della glicemia, chetoacidosi, allucinazioni, ecc. Il tasso di mortalità nei bambini è piuttosto elevato; negli adulti, senza trattamento, possono verificarsi coma e morte a causa di edema cerebrale.
Albinismo e alcaptonuria sono "uniti" solo dalla tirosina. L'albinismo, inclusa l'oculocutanea, è causato da mutazioni genetiche che influenzano la produzione del pigmento melanina. Alterazioni congenite sono state osservate nel gene TYR sul cromosoma 11 (11q14.3), che codifica per la tirosinasi, un enzima melanosomiale contenente rame necessario per la formazione del pigmento cutaneo a partire dai prodotti del metabolismo della tirosina. Questa malattia è molto più comune dell'alcaptonuria.
Complicazioni e conseguenze
Causate dall'azione dei metaboliti intermedi della tirosina – acido omogentisico e benzochinoneacetico – le conseguenze e le complicazioni dell'alcaptonuria si manifestano a causa della deposizione di polimeri pigmentati reattivi, della distruzione delle fibrille di collagene e del deterioramento delle condizioni della cartilagine (con diminuzione della loro resistenza allo stress meccanico).
Nel corso degli anni, in età adulta, si sviluppano artrite degenerativa e osteoartrite delle grandi articolazioni (anca, sacroiliaca e ginocchio); gli spazi intervertebrali si restringono (soprattutto nella colonna lombare e toracica) – con calcificazione e formazione di osteofiti; la densità del tessuto delle placche ossee subcondrali diminuisce e le ossa sottostanti possono subire un rimodellamento patologico con formazione di escrescenze e deformazioni. [ 11 ]
A causa della stessa calcificazione si possono osservare danni alle valvole cardiache (aortica e mitrale) e alle arterie coronarie – con segni di cardiopatia coronarica, nonché la formazione di calcoli nei reni e nella prostata. [ 12 ], [ 13 ]
Diagnostica alcaptonuria
Solitamente la diagnosi dei disturbi metabolici congeniti si basa sullo studio dei fluidi biologici del corpo.
In base a quali test e reazioni è possibile diagnosticare l'alcaptonuria? Sono necessari esami delle urine per rilevare l'acido omogentisinico e determinarne il livello (normale: 20-30 mg al giorno, elevato: 3-8 g). Un campione di urina viene esaminato mediante gascromatografia o spettrometria di massa, utilizzando la cromatografia liquida; è possibile un test di screening per la presenza di cloruro di ferro nelle urine. [ 14 ]
Esiste anche un metodo per una diagnosi rapida: la determinazione dell'alcapton nelle macchie di urina essiccata sulla carta (in base all'intensità del colore).
Per chiarire la diagnosi, la diagnostica strumentale (radiografia) consiste nell'identificare i segni radiologici dell'osteoartrite e di altre patologie articolari nei pazienti.
La diagnosi viene confermata da metodi genetici molecolari di diagnosi delle malattie ereditarie, come i test genetici e il sequenziamento del DNA. [ 15 ]
Diagnosi differenziale
La diagnosi differenziale comprende l'emocromatosi e l'insufficienza epatica acuta del neonato, la melaninuria, la porfiria acuta intermittente, la linfoistiocitosi emofagocitica, la patologia mitocondriale primaria, l'artrite reumatoide, la spondilite anchilosante.
Chi contattare?
Trattamento alcaptonuria
Il trattamento principale per l'alcaptonuria è la somministrazione orale di dosi elevate (almeno 1000 mg al giorno) di acido ascorbico. Nei bambini, ciò aumenta l'escrezione di acido omogentisico nelle urine e, negli adulti, riduce il contenuto urinario del suo derivato, l'acido benzochinoneacetico, e ne rallenta il legame alle strutture del tessuto connettivo delle articolazioni e al collagene. [ 16 ]
Cliniche dell'Europa occidentale stanno testando il farmaco Nitisinone (Orfalin), un farmaco appartenente al gruppo dei metaboliti che inibisce la seconda fase del catabolismo della tirosina: la trasformazione del para-idrossifenilpiruvato in acido omogentisico. Tuttavia, l'uso di questo agente farmacologico porta all'accumulo di tirosina e può causare gravi effetti collaterali, tra cui opacità corneale e fotofobia, epistassi e sanguinamento gastrico, insufficienza epatica, alterazioni ematiche, ecc. Ciononostante, negli Stati Uniti, il Nitisinone è approvato dalla FDA per il trattamento della tirosinemia di tipo I. [17 ], [ 18 ]
Per i problemi articolari causati dall'alcaptonuria si ricorre alla fisioterapia (terapia con esercizi per aumentare la forza muscolare e migliorare la mobilità articolare, balneoterapia e terapia con peloidi per limitare il dolore).
Sebbene la tirosina non venga solo fornita dagli alimenti ma anche prodotta dall'organismo, ai pazienti affetti da alcaptonuria si raccomanda di seguire una dieta povera di proteine e di limitare il consumo di alimenti ricchi di tirosina, principalmente carne di manzo e di maiale, latticini (in particolare formaggi), legumi, noci e semi.
Prevenzione
La prevenzione delle mutazioni genetiche è impossibile, ma per prevenire la nascita di bambini ad alto rischio di malattie congenite, esiste la consulenza genetica medica, necessaria prima di una gravidanza pianificata per quelle coppie la cui storia familiare comprende malattie ereditarie. [ 19 ]
Previsione
Gli esiti fatali dell'alcaptonuria sono molto rari e il decesso può essere causato da gravi complicazioni a carico di cuore e reni. Pertanto, l'aspettativa di vita complessiva delle persone affette da alcaptonuria è buona.
Ma la qualità della vita si riduce a causa di dolori intensi alle articolazioni o alla colonna vertebrale, con significativa limitazione della mobilità, spesso progressiva.