Esperto medico dell'articolo

Nuove pubblicazioni
Fibrosi cistica
Ultima recensione: 04.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
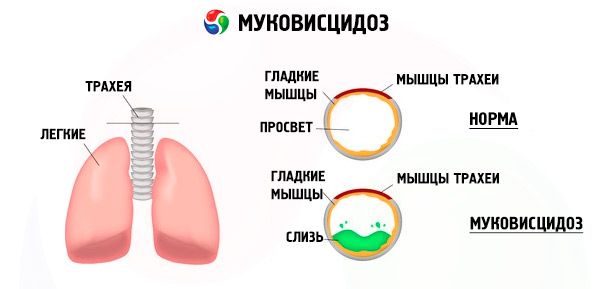
La fibrosi cistica è una malattia monogenica autosomica recessiva genetica caratterizzata da un disturbo della secrezione delle ghiandole esocrine degli organi vitali, con danni principalmente all'apparato respiratorio e digerente, decorso grave e prognosi sfavorevole.
 [ 1 ]
[ 1 ]
Epidemiologia
L'incidenza della fibrosi cistica oscilla tra 1:2.500 e 1:4.600 neonati. Ogni anno, in tutto il mondo nascono circa 45.000 persone con fibrosi cistica. L'incidenza dei portatori del gene della fibrosi cistica è del 3-4%, con circa 275 milioni di persone in tutto il mondo portatrici di questo gene, di cui circa 5 milioni vivono in Russia e circa 12,5 milioni nei paesi della CSI.
Le cause fibrosi cistica
La fibrosi cistica è trasmessa con modalità autosomica recessiva. Il gene della fibrosi cistica è localizzato nell'autosoma 7, contiene 27 esoni ed è costituito da 250.000 coppie di nucleotidi.
Un singolo gene può presentare numerose mutazioni, ciascuna delle quali è specifica di una particolare popolazione o regione geografica. Sono state descritte più di 520 mutazioni, la più comune delle quali è la delta-P-508, ovvero una sostituzione dell'amminoacido fenilalanina in posizione 508.
Patogenesi
Mutazioni nel gene della fibrosi cistica interrompono la struttura e la funzione di una proteina chiamata CFTR (regolatore transmembrana della fibrosi cistica). Questa proteina agisce come un canale del cloro coinvolto nello scambio acqua-elettroliti delle cellule epiteliali del sistema broncopolmonare, del tratto gastrointestinale, del pancreas, del fegato e dell'apparato riproduttivo. A seguito dell'interruzione della funzione e della struttura della proteina CFTR, gli ioni cloruro Cl- si accumulano all'interno della cellula. Ciò porta a una variazione del potenziale elettrico nel lume dei dotti escretori, che facilita il flusso di grandi quantità di ioni sodio (Na + ) dal lume del dotto verso la cellula e un ulteriore aumento dell'assorbimento di acqua dallo spazio pericellulare.
Come conseguenza di questi cambiamenti, la secrezione della maggior parte delle ghiandole esocrine si ispessisce, la sua evacuazione viene interrotta, il che porta a gravi disturbi secondari negli organi e nei sistemi, più pronunciati nell'apparato broncopolmonare e in quello digerente.
Nei bronchi si sviluppa un processo infiammatorio cronico di intensità variabile, la funzionalità dell'epitelio ciliato viene drasticamente compromessa, l'espettorato diventa molto viscoso, denso, molto difficile da evacuare, si osserva il suo ristagno, si formano bronchiolo- e bronchiectasie, che col tempo diventano più frequenti. Questi cambiamenti portano a un aumento dell'ipossia e alla formazione di cardiopatia polmonare cronica.
I pazienti con fibrosi cistica sono estremamente predisposti allo sviluppo di infiammazione cronica del sistema broncopolmonare. Ciò è dovuto a marcate alterazioni del sistema di difesa broncopolmonare locale (riduzione dei livelli di IgA, interferone, funzione fagocitaria dei macrofagi alveolari e dei leucociti).
I macrofagi alveolari svolgono un ruolo fondamentale nello sviluppo dell'infiammazione cronica nel sistema broncopolmonare. Producono elevate quantità di IL-8, che aumenta drasticamente la chemiotassi dei neutrofili nell'albero bronchiale. I neutrofili si accumulano in grandi quantità nei bronchi e, insieme alle cellule epiteliali, secernono numerose citochine proinfiammatorie, tra cui IL-1, IL-8, IL-6, fattore di necrosi tumorale e leucotrieni.
Un ruolo importante nella patogenesi del danno al sistema broncopolmonare è svolto anche dall'elevata attività dell'enzima elastasi. Si distingue tra elastasi esogena ed endogena. La prima è prodotta dalla flora batterica (in particolare Pseudomonas aeruginosa), la seconda dai leucociti neutrofili. L'elastasi distrugge l'epitelio e altri elementi strutturali dei bronchi, contribuendo a un'ulteriore interruzione del trasporto mucociliare e alla rapida formazione di bronchiectasie.
I leucociti neutrofili secernono anche altri enzimi proteolitici. L'alfa-1-antipirsina e l'inibitore secretorio delle leucoproteasi contrastano l'azione degli enzimi proteolitici e, quindi, proteggono il sistema broncopolmonare dai loro effetti dannosi. Tuttavia, purtroppo, nei pazienti con fibrosi cistica, questi fattori protettivi sono soppressi da una quantità significativa di proteasi neutrofila.
Tutte queste circostanze contribuiscono all'introduzione dell'infezione nel sistema broncopolmonare e allo sviluppo di bronchite purulenta cronica. Inoltre, va tenuto presente che la proteina difettosa codificata dal gene della fibrosi cistica altera lo stato funzionale dell'epitelio bronchiale, favorendo l'adesione di batteri, principalmente Pseudomonas aeruginosa, all'epitelio bronchiale.
Oltre alla patologia del sistema broncopolmonare, la fibrosi cistica provoca anche gravi danni al pancreas, allo stomaco, all'intestino crasso e tenue e al fegato.
Sintomi fibrosi cistica
La fibrosi cistica si manifesta con vari sintomi clinici. Nei neonati, la malattia può manifestarsi con ileo da meconio. A causa della mancanza o addirittura dell'assenza completa di tripsina, il meconio diventa molto denso e viscoso e si accumula nella regione ileocecale. Inoltre, si sviluppa un'ostruzione intestinale, che si manifesta con vomito intenso con aggiunta di bile, distensione addominale, mancata escrezione di meconio, sviluppo di sintomi peritonitici e rapido aumento delle manifestazioni cliniche della sindrome da intossicazione grave. Il bambino può morire nei primi giorni di vita se non viene eseguito un intervento chirurgico d'urgenza.
Nei casi meno gravi, un segno caratteristico della fibrosi cistica è l'emissione di feci abbondanti e frequenti, untuose, con una grande quantità di grasso e dall'odore molto sgradevole. In 1/3 dei pazienti si osserva prolasso del retto.
Successivamente, i pazienti continuano a manifestare disfunzioni intestinali, sindrome da malassorbimento, gravi disturbi dello sviluppo fisico e grave ipovitaminosi.
Nel primo o secondo anno di vita compaiono sintomi di danno al sistema broncopolmonare (forma lieve della malattia), che si manifestano con una tosse che può essere estremamente pronunciata e assomigliare a una tosse con pertosse. La tosse è accompagnata da cianosi, mancanza di respiro e separazione di espettorato denso, inizialmente mucoso e poi purulento. Gradualmente, si forma un quadro clinico di bronchite cronica ostruttiva e bronchiectasie, enfisema polmonare e insufficienza respiratoria. I bambini sono estremamente suscettibili alle infezioni respiratorie acute virali e batteriche, che contribuiscono alle riacutizzazioni e alla progressione della patologia broncopolmonare. È possibile lo sviluppo di asma bronchiale infezione-dipendente.
Nei bambini in età scolare, la fibrosi cistica può manifestarsi come "colica intestinale". I pazienti lamentano forti dolori addominali parossistici, gonfiore e vomito ripetuto. Alla palpazione dell'addome, si riscontrano formazioni dense, localizzate nella proiezione dell'intestino crasso: masse fecali miste a muco denso e denso. I bambini sono molto predisposti allo sviluppo di alcalosi ipocloremica a causa dell'eccessiva escrezione di sale con il sudore nella stagione calda, mentre sulla pelle del bambino compare una "brina salina".
Disturbi del sistema broncopolmonare negli adulti
Il danno al sistema broncopolmonare nei pazienti con fibrosi cistica (forma polmonare della malattia) è caratterizzato dallo sviluppo di bronchite ostruttiva purulenta cronica, bronchiectasie, polmonite cronica, enfisema polmonare, insufficienza respiratoria e cardiopatia polmonare. Alcuni pazienti sviluppano pneumotorace e altre complicanze della fibrosi cistica: atelettasia, ascessi polmonari, emottisi, emorragia polmonare e asma bronchiale infettivo.
I pazienti lamentano una tosse parossistica dolorosa con espettorato mucopurulento molto viscoso, difficile da separare, a volte con presenza di sangue. Inoltre, la mancanza di respiro è estremamente caratteristica, inizialmente durante lo sforzo fisico e poi a riposo. La mancanza di respiro è causata da ostruzione bronchiale. Molti pazienti lamentano rinite cronica causata da poliposi e sinusite. Sono caratteristici anche una significativa debolezza, un progressivo calo delle prestazioni e frequenti malattie virali respiratorie acute. All'esame obiettivo, l'attenzione viene attirata da pallore cutaneo, gonfiore del viso, cianosi delle mucose visibili e grave mancanza di respiro. Con lo sviluppo di una cardiopatia polmonare scompensata, compare edema alle gambe. Si può osservare un ispessimento delle falangi terminali delle dita a forma di bacchette di tamburo e delle unghie a forma di vetri da orologio. Il torace assume una forma a botte (a causa dello sviluppo di enfisema polmonare).
La percussione polmonare rivela segni di enfisema: un suono a scatola, una netta limitazione della mobilità del margine polmonare e un abbassamento del margine inferiore dei polmoni. L'auscultazione polmonare rivela respiro affannoso con espirazione prolungata, respiro sibilante secco e sparso, respiro sibilante umido, medio e fine, con gorgoglii. In caso di enfisema polmonare grave, la respirazione è notevolmente indebolita.
Manifestazioni extrapolmonari della fibrosi cistica
Le manifestazioni extrapolmonari della fibrosi cistica possono essere piuttosto pronunciate e verificarsi frequentemente.
Danno pancreatico
Un'insufficienza della funzione esocrina del pancreas di varia gravità è osservata nell'85% dei pazienti con fibrosi cistica. In caso di danno pancreatico lieve, sono assenti sindromi da maldigestione e malassorbimento; si osservano solo manifestazioni di laboratorio dell'insufficienza esocrina (bassi livelli di tripsina e lipasi nel sangue e nel contenuto duodenale; spesso grave steatorrea). È noto che per prevenire la sindrome da maldigestione è sufficiente una secrezione pari anche solo all'1-2% della lipasi totale. Solo i disturbi significativi della funzione esocrina si manifestano clinicamente.
In condizioni normali, gli acini del pancreas producono una secrezione liquida ricca di enzimi. Man mano che la secrezione scorre lungo il dotto escretore, si arricchisce di acqua e anioni, diventando ancora più liquida. Nella fibrosi cistica, a causa di un disturbo nella struttura e nella funzione del regolatore transmembrana (canale del cloro), la secrezione pancreatica non riceve una quantità sufficiente di liquido, diventa viscosa e la velocità del suo movimento lungo il dotto escretore rallenta drasticamente. Le proteine della secrezione si depositano sulle pareti dei piccoli dotti escretori, causandone l'ostruzione. Con il progredire della malattia, si sviluppano infine la distruzione e l'atrofia degli acini: si forma una pancreatite cronica con insufficienza pancreatica esocrina. Ciò si riflette clinicamente nello sviluppo di sindromi da maldigestione e malassorbimento. L'insufficienza pancreatica è la causa principale del malassorbimento dei grassi nella fibrosi cistica, ma questo si osserva solitamente in presenza di un significativo deficit di lipasi. Forsher e Durie (1991) indicano che in completa assenza di lipasi pancreatica, i grassi vengono scomposti e assorbiti al 50-60%, il che è dovuto alla presenza di lipasi gastriche e salivari (sublinguali), la cui attività è prossima al limite inferiore della norma. Insieme all'interruzione della scomposizione e dell'assorbimento dei grassi, si verifica anche un'interruzione della scomposizione e del riassorbimento delle proteine. Circa il 50% delle proteine assunte con il cibo viene perso con le feci. L'assorbimento dei carboidrati è influenzato in misura minore nonostante la carenza di α-amilasi, ma il metabolismo dei carboidrati può essere significativamente alterato.
Il danno al pancreas si manifesta con lo sviluppo di una sindrome da maldigestione e malassorbimento, con significativa perdita di peso e abbondanti feci grasse.
Lo sviluppo di sindromi da maldigestione e malassorbimento è facilitato anche da una grave disfunzione delle ghiandole intestinali, da una secrezione alterata dei succhi intestinali e da una diminuzione del contenuto di enzimi intestinali in essi.
Le sindromi da maldigestione e malassorbimento sono anche chiamate forma intestinale della fibrosi cistica.
Nei pazienti con fibrosi cistica, nelle fasi avanzate della malattia (nel 2% dei bambini e nel 15% degli adulti), si osserva una compromissione della funzione endocrina del pancreas (diabete mellito).
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Danni al fegato e alle vie biliari
Nel 13% dei pazienti con forme miste e intestinali di fibrosi cistica si sviluppa cirrosi epatica. È più tipica per le mutazioni W128X, delta-P508 e X1303K. La cirrosi biliare epatica con ipertensione portale viene riscontrata nel 5-10% dei pazienti. Secondo Welch, Smith (1995), segni clinici, morfologici, di laboratorio e strumentali di danno epatico vengono rilevati nell'86% dei pazienti con fibrosi cistica.
Molti pazienti affetti da fibrosi cistica sviluppano anche una colecistite cronica, spesso calcolosa.
Disfunzione delle ghiandole sessuali
I pazienti con fibrosi cistica possono presentare azoospermia, che è la causa dell'infertilità. La ridotta fertilità è tipica anche delle donne.
Fasi
Esistono tre gradi di gravità della fibrosi cistica polmonare.
La forma lieve della fibrosi cistica è caratterizzata da rare riacutizzazioni (non più di una volta all'anno); durante i periodi di remissione, le manifestazioni cliniche sono praticamente assenti e i pazienti sono in grado di lavorare.
Gravità moderata: le riacutizzazioni si osservano 2-3 volte l'anno e durano circa 2 mesi o più. Nella fase di riacutizzazione, si manifestano tosse intensa con espettorato difficile da separare, mancanza di respiro anche con sforzi fisici minimi, temperatura corporea subfebbrile, debolezza generale e sudorazione. Contemporaneamente, si verifica una compromissione della funzione esocrina del pancreas. Nella fase di remissione, la capacità lavorativa non viene completamente ripristinata e persiste la mancanza di respiro durante lo sforzo fisico.
Il decorso grave è caratterizzato da riacutizzazioni molto frequenti della malattia. Le remissioni sono praticamente assenti. Il quadro clinico è caratterizzato da grave insufficienza respiratoria, sintomi di cardiopatia polmonare cronica, spesso scompensati, con emottisi tipica. Si osserva una significativa perdita di peso e i pazienti sono completamente invalidi. Di norma, la broncopneumopatia grave è accompagnata da una disfunzione pancreatica nettamente marcata.
Forme
- Lesioni broncopolmonari
- Polmonite ripetuta e ricorrente con decorso prolungato.
- Polmonite ascessuale, soprattutto nei neonati.
- Polmonite cronica, soprattutto bilaterale.
- Asma bronchiale refrattaria alla terapia tradizionale.
- Bronchite ricorrente, bronchiolite, soprattutto con coltura di Pseudomonas aeruginosa.
- Cambiamenti nel tratto gastrointestinale
- Ileo da meconio e suoi equivalenti.
- Sindrome da alterato assorbimento intestinale di genesi sconosciuta.
- Ittero ostruttivo nei neonati con decorso prolungato.
- Cirrosi epatica.
- Diabete mellito.
- Reflusso gastroesofageo.
- Colelitiasi.
- Prolasso rettale.
- Cambiamenti in altri organi e sistemi
- Disturbi della crescita e dello sviluppo.
- Ritardo nello sviluppo sessuale.
- Infertilità maschile.
- Polipi nasali.
- Fratelli e sorelle provenienti da famiglie con fibrosi cistica.
[ 24 ]
Complicazioni e conseguenze
Le complicazioni del tratto gastrointestinale includono:
- Il diabete mellito si sviluppa nell'8-12% dei pazienti di età superiore ai 25 anni.
- Colonpatia fibrosante.
- Ileo da meconio nel periodo neonatale (nel 12% dei neonati affetti da fibrosi cistica), sindrome da ostruzione intestinale distale, prolasso rettale, ulcera peptica e malattia da reflusso gastroesofageo.
Complicanze epatiche:
- Malattia del fegato grasso (nel 30-60% dei pazienti),
- Cirrosi biliare focale, cirrosi biliare multinodulare e ipertensione portale associata.
L'ipertensione portale a volte può portare alla morte a causa delle varici esofagee.
La prevalenza di colecistite e calcoli biliari è più elevata nei pazienti affetti da fibrosi cistica rispetto ad altri individui.
Pubertà ritardata, riduzione della fertilità e altre complicazioni. La maggior parte degli uomini presenta azoospermia e iposviluppo dei vasi deferenti.
Diagnostica fibrosi cistica
Analisi del sangue generali: l'anemia di varia gravità è tipica, solitamente normocromica o ipocromica. L'anemia ha una genesi polifattoriale (ridotto assorbimento di ferro e vitamina B12 nell'intestino a causa dello sviluppo di una sindrome da malassorbimento). È possibile leucopenia, con sviluppo di bronchite purulenta e polmonite - leucocitosi, aumento della VES.
Analisi generale delle urine: nessuna variazione significativa, in rari casi si osserva una leggera proteinuria.
Esame coprologico: si osservano steatorrea e creatorrea. Becker (1987) raccomanda la misurazione della chimotripsina e degli acidi grassi nelle feci. Prima di determinare la chimotripsina nelle feci, è necessario interrompere l'assunzione di enzimi digestivi almeno 3 giorni prima dell'esame. Nella fibrosi cistica, la quantità di chimotripsina nelle feci è ridotta e la quantità di acidi grassi è aumentata (l'escrezione normale di acidi grassi è inferiore a 20 mmol/die). È necessario tenere presente che un'aumentata escrezione di acidi grassi con le feci si osserva anche in:
- carenza di acidi grassi coniugati nell'intestino tenue dovuta a insufficienza epatica, ostruzione dei dotti biliari, significativa colonizzazione batterica dell'intestino tenue (in questo caso si verifica un'intensa idrolisi degli acidi biliari);
- ileite;
- celiachia (con sviluppo di sindrome da malassorbimento);
- enterite;
- linfomi intestinali;
- Malattia di Whipple;
- allergie alimentari;
- transito accelerato di masse alimentari nella diarrea di varia origine, sindrome carcinoide, tireotossicosi.
Esame biochimico del sangue: diminuzione dei livelli di proteine totali e albumina, aumento dei livelli di alfa2 e gammaglobuline, bilirubina e aminotransferasi (in caso di danni al fegato), diminuzione dell'attività di amilasi, lipasi, tripsina e livelli di ferro e calcio (in caso di sviluppo di sindrome da maldigestione, malassorbimento).
Analisi dell'espettorato: presenza di un gran numero di leucociti neutrofili e microrganismi (durante la batterioscopia dell'espettorato).
Uno studio sulla funzione di assorbimento dell'intestino tenue e sulla funzione esocrina del pancreas rivela disturbi significativi.
Esame radiografico dei polmoni: rivela alterazioni, la cui gravità dipende dalla gravità e dalla fase della malattia. Le alterazioni più caratteristiche sono:
- aumento del pattern polmonare dovuto a cambiamenti interstiziali peribronchiali;
- espansione delle radici dei polmoni;
- quadro di atelettasia lobulare, subsegmentale o anche segmentale dei polmoni;
- aumento della trasparenza dei campi polmonari, soprattutto nelle sezioni superiori, posizione bassa e mobilità insufficiente del diaframma, dilatazione dello spazio retrosternale (manifestazione di enfisema polmonare);
- infiltrazione segmentale o polisegmentale del tessuto polmonare (nello sviluppo della polmonite).
La broncografia rivela alterazioni causate dall'ostruzione dei bronchi da parte di espettorato viscoso (frammentazione del riempimento dei bronchi con contrasto, contorni irregolari, fenomeno di rottura bronchiale, significativa diminuzione del numero di rami laterali), nonché broncoecgas (cilindriche, miste), localizzate soprattutto nelle parti inferiori dei polmoni.
La broncoscopia rivela una bronchite purulenta diffusa con abbondante espettorato denso e viscoso e pellicole fibrinose.
Spirometria - già nelle fasi iniziali della malattia rivela un'insufficienza respiratoria di tipo ostruttivo (riduzione di FVC, FEV1, indice di Tiffno), restrittivo (riduzione di FVC) o, più spesso, ostruttivo-restrittivo (riduzione di FVC, FVC, FEV1, indice di Tiffno).
Il test del sudore di Gibson e Cook (test degli elettroliti del sudore) prevede la stimolazione della sudorazione mediante elettroforesi della pilocarpina con successiva determinazione dei cloruri nel sudore. Doerehuk (1987) descrive il test come segue. L'elettroforesi della pilocarpina viene eseguita sull'avambraccio, con una corrente elettrica di 3 mA. Dopo aver deterso la pelle con acqua distillata, il sudore viene raccolto utilizzando carta da filtro posizionata sulla zona stimolata, coperta con una garza per impedirne l'evaporazione. Dopo 30-60 minuti, la carta da filtro viene rimossa e diluita in acqua distillata. Viene misurata la quantità di sudore raccolto. Per ottenere risultati affidabili, è necessario raccogliere almeno 50 mg (preferibilmente 100 mg) di sudore.
Se la concentrazione di cloruro è superiore a 60 mmol/l, la diagnosi di fibrosi cistica è considerata probabile; se la concentrazione di cloruro è superiore a 100 mmol/l, è affidabile; in questo caso, la differenza tra la concentrazione di cloro e sodio non dovrebbe superare 8-10 mmol/l. Hadson (1983) raccomanda che se il contenuto di sodio e cloruro nel sudore è borderline, venga eseguito un test al prednisolone (5 mg per via orale per 2 giorni, seguito dalla determinazione degli elettroliti nel sudore). Negli individui che non soffrono di fibrosi cistica, il livello di sodio nel sudore diminuisce fino al limite inferiore della norma; nella fibrosi cistica, non cambia. Un test del sudore è raccomandato per ogni bambino con tosse cronica.
L'analisi di campioni di sangue o di DNA per le principali mutazioni del gene della fibrosi cistica è il test diagnostico più sensibile e specifico. Tuttavia, questo metodo è adatto solo ai Paesi in cui il tasso di mutazione delta-P508 è superiore all'80%. Inoltre, la tecnica è molto costosa e tecnicamente complessa.
La diagnosi prenatale della fibrosi cistica si esegue determinando gli isoenzimi della fosfatasi alcalina nel liquido amniotico. Questo metodo è possibile a partire dalla 18a-20a settimana di gravidanza.
I criteri principali per la diagnosi della fibrosi cistica sono i seguenti:
- indicazioni nell'anamnesi di ritardo dello sviluppo fisico nell'infanzia, malattie respiratorie croniche ricorrenti, disturbi dispeptici e diarrea, presenza di fibrosi cistica nei parenti stretti;
- bronchite cronica ostruttiva, spesso ricorrente, con sviluppo di bronchiectasie ed enfisema polmonare, spesso polmonite ricorrente;
- pancreatite cronica ricorrente con marcata diminuzione della funzione esocrina, sindrome da malassorbimento;
- aumento del contenuto di cloro nel sudore del paziente;
- infertilità con funzione sessuale preservata.
L'identificazione dei gruppi a rischio facilita la diagnosi efficace e la diagnosi differenziale della fibrosi cistica.
Programma di screening per la fibrosi cistica
- Analisi generali del sangue, delle urine, dell'espettorato.
- Analisi batteriologica dell'espettorato.
- Analisi coprologica.
- Esame biochimico del sangue: determinazione delle proteine totali e delle frazioni proteiche, glucosio, bilirubina, aminotransferasi, fosfatasi alcalina, gamma-glutamil transpeptidasi, potassio, calcio, ferro, lipasi, amilasi, tripsina.
- Studio della funzione esocrina del pancreas e della funzione assorbente dell'intestino.
- Fluoroscopia e radiografia dei polmoni, TAC dei polmoni.
- Elettrocardiogramma.
- Ecocardiografia.
- Broncoscopia e broncografia.
- Spirometria.
- Test del sudore.
- Consultazione con un genetista.
- Analisi di gocce di sangue o campioni di DNA per individuare le principali mutazioni del gene della fibrosi cistica.
Quali test sono necessari?
Chi contattare?
Trattamento fibrosi cistica
Il tipo e la gravità dei sintomi della fibrosi cistica possono variare notevolmente, pertanto non esiste un piano di trattamento standard: è personalizzato per ogni individuo.
La terapia consiste nelle seguenti misure terapeutiche:
- Esercizi di respirazione e drenaggio posturale aiutano a eliminare il muco denso che si accumula nei polmoni. Alcune tecniche di disostruzione delle vie aeree richiedono l'aiuto di familiari, amici o di uno pneumologo. Molte persone utilizzano un giubbotto gonfiabile che vibra ad alta frequenza.

- Farmaci inalatori che hanno effetti broncodilatatori, drenanti (mucolitici) e antibatterici (ad esempio, fluorochinoloni).
- Preparati contenenti enzimi pancreatici per migliorare la digestione. Questi preparati vengono assunti durante i pasti.
- Multivitaminici (comprese le vitamine liposolubili).
Nel 2015, la FDA ha approvato un secondo farmaco per il trattamento della fibrosi cistica che agisce su una proteina difettosa nota come CFTR. Il primo farmaco, un cosiddetto modulatore del CFTR, è stato approvato nel 2012. Si prevede che i modulatori del CFTR possano prolungare la vita di alcune persone affette da fibrosi cistica di decenni.
Potrebbe essere necessario un intervento chirurgico per trattare le seguenti complicazioni respiratorie:
- Pneumotorace, emottisi massiva ricorrente o persistente, polipi nasali, sinusite persistente e cronica.
- Ileo da meconio, intussuscezione, prolasso rettale.
Il trapianto polmonare viene eseguito nella fase terminale della malattia.
Previsione
L'età media di sopravvivenza dei pazienti con fibrosi cistica varia dai 35 ai 40 anni. L'età media di sopravvivenza è più alta negli uomini che nelle donne.
Con le moderne strategie terapeutiche, l'80% dei pazienti raggiunge l'età adulta. Tuttavia, la fibrosi cistica limita significativamente le capacità funzionali del paziente. Non esiste ancora una cura per questa malattia.