Esperto medico dell'articolo

Nuove pubblicazioni
Xeroderma pigmentoso
Ultima recensione: 04.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
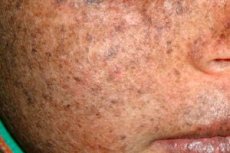
Lo xeroderma pigmentoso è una malattia genetica autosomica recessiva che causa la riparazione del DNA. La malattia è causata da mutazioni nei geni coinvolti nella riparazione del DNA danneggiato dalle radiazioni ultraviolette e da altre sostanze tossiche.
Questa malattia colpisce più spesso i bambini, chiamati anche "bambini della notte". Le complicanze più frequenti sono il carcinoma basocellulare e altre neoplasie maligne della pelle, il melanoma maligno metastatico e il carcinoma squamocellulare.
 [ 1 ]
[ 1 ]
Le cause xeroderma pigmentoso
Patogenesi
La carenza o la completa assenza degli enzimi endonucleasi UV nelle cellule del paziente (nei fibroblasti) gioca un ruolo importante nello sviluppo della malattia. Questo enzima è responsabile della riproduzione del DNA danneggiato dai raggi ultravioletti. Secondo altre fonti, la carenza dell'enzima DNA polimerasi-1 gioca un ruolo importante nello sviluppo della malattia in alcuni pazienti. La malattia si sviluppa più spesso sotto l'influenza di raggi con una lunghezza d'onda di 280-310 nm. Si ritiene che la xerodermia pigmentaria si sviluppi a causa dell'ingresso di varie sostanze fotodinamiche nell'organismo o di un aumento delle porfirine nell'ambiente biologico umano.
Nelle fasi iniziali della malattia si osservano ipercheratosi, atrofia del focolaio epidermico, assottigliamento dello strato malpighiano, aumento dei granuli di melanina nello strato basale delle cellule e infiltrazione infiammatoria cronica nello strato superiore del derma (principalmente intorno ai vasi sanguigni). Successivamente, si osservano alterazioni degenerative del collagene e delle fibre elastiche e, allo stadio tumorale, si manifestano i segni caratteristici del cancro della pelle.
Sintomi xeroderma pigmentoso
Uomini e donne sono colpiti in egual misura da questa malattia. La malattia inizia nella prima infanzia, in primavera o in estate. La pelle del paziente è particolarmente sensibile alla luce solare. Pertanto, la prima eruzione cutanea, sotto forma di iperpigmentazione, simile a un neo, compare sulle aree cutanee esposte alla luce solare. L'eruzione cutanea peggiora, l'eritema si intensifica, assume un colore marrone scuro, compaiono teleangectasie, angiomi e atrofia. Successivamente, si sviluppano papillomi e verruche (principalmente sulla pelle del viso e del collo), macchie e ulcere. A causa delle cicatrici delle ulcere, il naso si assottiglia ("a becco d'uccello") e la palpebra si estroflette. I papillomi di solito si sviluppano in tumori maligni. Lo xeroderma pigmentato si manifesta solitamente insieme al carcinoma spinocellulare e ai melanosarcomi.
Cosa ti infastidisce?
Fasi
Nel decorso clinico dello xeroderma pigmentoso si distinguono cinque stadi: infiammatorio (eritematoso), iperpigmentazione, atrofia, ipercheratosi e tumori maligni.
Durante la fase infiammatoria (eritematosa), compaiono gonfiore, macchie rosse e talvolta vesciche e vescicole sulle aree cutanee esposte alla luce solare (viso, collo, parte superiore del torace, braccia, mani). Non si osserva quasi nessuna eruzione cutanea nelle aree non esposte alla luce solare.
In caso di iperpigmentazione, al posto delle macchie rosse compaiono delle macchie iperpigmentate color marrone chiaro, simili a nei.
Con l'atrofia, la pelle si secca, si assottiglia e si formano le rughe. Sulla pelle delle labbra e del naso si osservano numerose piccole teleangectasie stellate e cicatrici con superficie lucida. A causa dell'atrofia e delle cicatrici, si sviluppano microtomia (riduzione dell'apertura della bocca), ectropion, assottigliamento di orecchie e naso, atresia dell'apertura nasale e della bocca. Nell'80-85% dei pazienti, gli occhi sono colpiti: si notano congiuntivite, cheratite, danni alla cornea e alle mucose e indebolimento della vista. Sulla pelle delle palpebre si osservano discromie, teleangectasie, alterazioni ipercheratosiche e tumori.
Con l'ipercheratosi, nei focolai patologici sopra descritti compaiono tumori verrucosi, papillomi, cheratoacantomi, fibromi, angiofibriomi e altri tumori benigni. Pertanto, alcuni scienziati includono la xerodermia pigmentaria nel gruppo delle malattie precancerose.
Dopo 10-15 anni dall'esordio della malattia, tumori cutanei maligni (basalioma, endotelioma, angiosarcoma) compaiono in focolai atroficamente alterati e macchie pigmentarie. I tumori vengono distrutti in breve tempo, metastatizzano agli organi interni e portano alla morte. Negli organi e nei tessuti interni di alcuni pazienti si osservano alterazioni distrofiche generalizzate (sindattilia del secondo e terzo dito del piede, distrofia dentale, perdita completa dei capelli, ecc.).
Forme
La forma neurologica dello xeroderma pigmentoso si manifesta in due sindromi.
La sindrome di Reed è caratterizzata da manifestazioni cliniche di xeroderma pigmentoso e microcefalia, idiopatia e lenta crescita del sistema scheletrico. Nella forma neurologica della malattia, la riparazione del DNA è difficoltosa e la radioterapia porta a un aumento dei principali focolai patologici.
La sindrome di De Sinctis-X Cocchione è caratterizzata dai seguenti segni clinici:
- sviluppo di segni clinici di xeroderma pigmentoso su pelli particolarmente fotosensibili;
- comparsa precoce di tumori maligni;
- decorso simultaneo di paralisi spastica, microcefalia e demenza;
- malformazioni congenite e nanismo;
- sottosviluppo delle gonadi;
- aborti spontanei frequenti;
- trasmissione recessiva per via ereditaria.
Secondo alcuni dermatologi, la sindrome di Santis-Cacchione non è una malattia indipendente, bensì una manifestazione clinica grave e pienamente espressa dello xeroderma pigmentoso.
Forme genetiche
Tipi |
Gene |
Luogo |
Descrizione |
Tipo A, I, XPA |
XPA |
9q22.3 |
Xeroderma pigmentoso (XP) gruppo A – forma classica |
Tipo B, II, XPB |
XPB |
2q21 |
Gruppo XP B |
Tipo C, III, XPC |
XPC |
3p25 |
Gruppo XP C |
Tipo D, IV, XPD |
XPD ERCC6 |
19q13.2-q13.3, 10q11 |
XP Gruppo D o Sindrome di De Sanctis-Cacchione |
Tipo E, V, XPE |
DDB2 |
11p12-p11 |
Gruppo XP E |
Tipo F, VI, XPF |
ERCC4 |
16p13.3-p13.13 |
Gruppo XP F |
Tipo G, VII, XPG |
RAD2ERCC5 |
13q33 |
XP gruppo G e sindrome COFS (sindrome cerebro-oculo-facio-scheletrica) tipo 3 |
Tipo V, XPV |
POLH |
6p21.1-p12 |
Variante di Xeroderma Pigmentosa |
Cosa c'è da esaminare?
Come esaminare?
Chi contattare?
Trattamento xeroderma pigmentoso
I farmaci antimalarici (delagyl, plaquenil, resoquin, ecc.) proteggono il DNA dai raggi ultravioletti, inibiscono il processo di depolimerizzazione e riducono la sensibilità (fotosensibilizzazione) della pelle alla luce solare. Questi farmaci hanno proprietà antinfiammatorie e iposensibilizzazione. Si raccomanda di associare la terapia generale a quella vitaminica (B1, B2, PP, B6, B12, A, E), antistaminici (tavegil, difenidramina, suprastin) e agenti desensibilizzanti (tiosolfato di sodio, cloruro di calcio al 10% per via endovenosa 10 ml).
Per il trattamento locale si utilizzano creme e unguenti solari.
Nella forma tumorale di xeroderma pigmentario, vengono utilizzati metodi chirurgici, azoto liquido e raggi laser. Per proteggersi dalla luce solare, è necessario indossare abiti comodi, cappelli da sole e guanti.
Previsione
La maggior parte dei pazienti (2/3) muore prima di raggiungere i 15 anni di età. Alcuni pazienti possono vivere fino a 40-50 anni.
[ 20 ]