Esperto medico dell'articolo

Nuove pubblicazioni
Encefalomiopatia di Leah subacuta necrotizzante
Ultima recensione: 04.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
La malattia è stata menzionata per la prima volta nel 1951. Ad oggi, ne sono stati descritti più di 120 casi. La malattia di Leigh (OMIM 256000) è una malattia geneticamente eterogenea che può essere ereditata per via nucleare (autosomica recessiva o legata al cromosoma X) o mitocondriale (meno comune).
 [
[ Le cause della sindrome di Leah
La malattia è causata da una carenza di enzimi che forniscono energia, dovuta principalmente a un'alterazione del metabolismo dell'acido piruvico e a un difetto nel trasporto degli elettroni nella catena respiratoria. Si sviluppa un deficit del complesso della piruvato deidrogenasi (subunità α-E1), della piruvato carbossilasi, del complesso 1 (NAD-coenzima Q-reduttasi) e del complesso 4 (citocromo ossidasi) della catena respiratoria.
È stato stabilito che i difetti della piruvato carbossilasi, del complesso 1 (NAD-coenzima Q-reduttasi) e del complesso 4 (citocromo ossidasi) della catena respiratoria sono ereditati con modalità autosomica recessiva, mentre i difetti del complesso della piruvato deidrogenasi (subunità α-E1) sono ereditati con modalità recessiva legata al cromosoma X. In caso di mutazioni puntiformi del mtDNA, che colpiscono la sesta subunità dell'ATPasi, l'ereditarietà mitocondriale è tipica. Più frequentemente si verifica una mutazione miscens, associata alla sostituzione della timina con guanina o citosina in posizione 8993 del mtDNA. Meno comune è una mutazione in posizione 9176 del mtDNA. Poiché la mutazione T8993G è il difetto principale nella sindrome NARP, sono state descritte famiglie con queste due patologie. Nei bambini è stata descritta anche una mutazione nel mtDNA in posizione 8344, che si verifica nella sindrome MERRF.
Si presume che l'accumulo di mtDNA mutante nella maggior parte dei mitocondri determini un decorso grave della sindrome di Leigh. Nella genesi mitocondriale di questa condizione, il mtDNA mutante è presente nel 90% di tutti i mitocondri. La patogenesi è associata a una compromissione della produzione di energia nelle cellule e allo sviluppo di acidosi lattica.
Sintomi della sindrome di Leah
I primi segni della malattia si manifestano in età precoce (1-3 anni). Tuttavia, sono noti casi di manifestazione della malattia a 2 settimane e a 6-7 anni di età. Inizialmente si sviluppano disturbi aspecifici: ritardo dello sviluppo psicomotorio, diminuzione dell'appetito, episodi di vomito, deficit del peso corporeo. Successivamente, i sintomi neurologici aumentano: ipotonia o distonia muscolare con transizione a ipertonia, attacchi di mioclono o crisi tonico-cloniche, tremore degli arti, coreoatetosi, disturbi della coordinazione, riduzione dei riflessi tendinei, letargia, sonnolenza. La neurodegenerazione cerebrale è progressiva. Aumentano i sintomi di insufficienza piramidale ed extrapiramidale, la deglutizione è compromessa. Si osservano spesso alterazioni dell'organo della vista come ptosi, oftalmoplegia, atrofia dei nervi ottici e, meno frequentemente, degenerazione pigmentaria della retina. Talvolta si sviluppa cardiomiopatia ipertrofica e compaiono episodi di tachipnea.
Raramente, la malattia si manifesta come un'encefalopatia acuta. Più tipico è il decorso cronico o subacuto, che porta a un esito fatale diversi anni dopo l'esordio della malattia. In caso di decorso rapido (diverse settimane), la morte sopraggiunge a causa della paralisi del centro respiratorio.
Diagnostica della sindrome di Leah
Un esame del sangue biochimico rivela acidosi lattica dovuta all'accumulo di acido lattico e piruvico nel sangue e nel liquido cerebrospinale, nonché un aumento del contenuto di alanina nel sangue. Anche il livello di corpi chetonici può essere elevato. Si rileva un'aumentata escrezione di acidi organici nelle urine: lattico, fumarico, ecc. Il livello di carnitina nel sangue e nei tessuti spesso diminuisce.
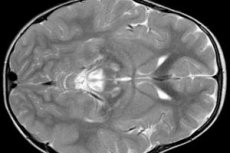
L'EEG rivela segni focali di attività epilettica. La risonanza magnetica rivela dilatazione dei ventricoli cerebrali, danno cerebrale bilaterale e calcificazione dei gangli della base (nucleo caudato, putamen, sostanza nera, globo pallido). È inoltre possibile rilevare atrofia degli emisferi cerebrali e della materia cerebrale.
L'esame morfologico rivela macroscopiche alterazioni della materia cerebrale: focolai simmetrici di necrosi, demielinizzazione e degenerazione spongiosa dell'encefalo, principalmente delle sezioni medie, del ponte, dei gangli della base, del talamo e del nervo ottico. Il quadro istologico include degenerazione cistica del tessuto cerebrale, gliosi astrocitaria, morte neuronale e aumento del numero di mitocondri nelle cellule. Nei muscoli scheletrici si osserva accumulo di inclusioni lipidiche, riduzione della reazione istochimica ai complessi 1 e 4 della catena respiratoria, accumulo subsarcolemmale di mitocondri, mitocondri anomali con disorganizzazione delle creste. Il fenomeno della RRF spesso non viene rilevato.
Come esaminare?
Quali test sono necessari?
Использованная литература