Esperto medico dell'articolo

Nuove pubblicazioni
Sindrome di Martin-Bell
Ultima recensione: 04.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
La sindrome di Martin-Bell fu descritta nel 1943 dai medici, da cui prese il nome. La malattia è un disturbo genetico che comporta ritardo mentale. Nel 1969, furono identificate alterazioni del cromosoma X (fragilità del braccio distale) caratteristiche di questa malattia. Nel 1991, gli scienziati hanno scoperto il gene responsabile dello sviluppo di questa malattia. Questa malattia è anche chiamata "sindrome dell'X fragile". Sia i ragazzi che le ragazze sono suscettibili alla malattia, ma i ragazzi sono più colpiti (tre volte di più).
Epidemiologia
La sindrome di Martin-Bell è una malattia piuttosto comune: ne soffrono 0,3-1,0 uomini su 1.000 e 0,2-0,6 donne su 1.000. Inoltre, i bambini con sindrome di Martin-Bell nascono in tutti i continenti con la stessa frequenza. Ovviamente, la nazionalità, il colore della pelle, la forma degli occhi, le condizioni di vita e il benessere delle persone non influenzano l'insorgenza della malattia. La sua frequenza è paragonabile solo a quella della sindrome di Down (1 caso ogni 600-800 neonati). Un quinto dei portatori maschi del gene alterato è sano, non presenta anomalie cliniche o genetiche, i restanti presentano segni di ritardo mentale da lieve a grave. Tra le portatrici femmine, poco più di un terzo è malato.
La sindrome dell'X fragile colpisce circa 1 uomo su 2.500-4.000 e 1 donna su 7.000-8.000. La prevalenza di portatori tra le donne è stimata in 1 su 130-250; la prevalenza di portatori tra gli uomini è stimata in 1 su 250-800.
Le cause Sindrome di Martin-Bell
La sindrome di Martin-Bell si sviluppa a causa della cessazione completa o parziale della produzione di una specifica proteina da parte dell'organismo. Ciò si verifica a causa della mancata risposta del gene FMR1, localizzato sul cromosoma X. La mutazione si verifica a seguito della ristrutturazione del gene a partire da varianti strutturali instabili degli stati genici (alleli), e non dall'inizio. La malattia si trasmette solo per via maschile e l'uomo non è necessariamente malato. I portatori maschi trasmettono il gene alle figlie in forma immodificata, quindi il loro ritardo mentale non è evidente. Con l'ulteriore trasmissione del gene dalla madre ai figli, il gene muta e compaiono tutti i segni caratteristici di questa malattia.
Patogenesi
La patogenesi della sindrome di Martin-Bell si basa su mutazioni dell'apparato genico, che portano al blocco della produzione della proteina FMR, una proteina vitale per l'organismo, soprattutto nei neuroni, e presente in vari tessuti. La ricerca dimostra che le proteine FMR sono direttamente coinvolte nei processi di regolazione delle traduzioni che avvengono nel tessuto cerebrale. L'assenza di questa proteina o la sua limitata produzione da parte dell'organismo porta al ritardo mentale.
Nella patogenesi della malattia, l'ipermetilazione genica è considerata un disturbo chiave, ma non è ancora stato possibile identificare in modo definitivo il meccanismo di sviluppo di questo disturbo.
Contemporaneamente, è stata scoperta anche l'eterogeneità del locus patologico, associata al poliallelismo, e al polilocus. È stata determinata la presenza di varianti alleliche nello sviluppo della malattia, causate dall'esistenza di mutazioni puntiformi e dalla distruzione del gene tipo FMRL.
I pazienti presentano anche 2 triplette fragili sensibili all'acido folico, localizzate a 300 kb, nonché 1,5-2 milioni di bp dalla tripletta fragile contenente il gene FMR1. Il meccanismo delle mutazioni che si verificano nei geni FRAXE e FRAXF (identificate nelle triplette fragili sopra menzionate) è correlato al meccanismo dei disturbi nella sindrome di Martin Bell. Questo meccanismo è causato dalla diffusione delle ripetizioni GCC e CGG, che causano la metilazione delle cosiddette isole CpG. Oltre alla forma classica della patologia, esistono anche 2 tipi rari che differiscono per l'espansione delle ripetizioni trinucleotidiche (nella meiosi maschile e femminile).
È stato scoperto che nella forma classica della sindrome, il paziente è privo di una speciale proteina nucleocitoplasmatica di tipo FMR1, che svolge la funzione di legare diversi mRNA. Inoltre, questa proteina promuove la formazione di un complesso che contribuisce allo svolgimento dei processi di traduzione all'interno dei ribosomi.
Sintomi Sindrome di Martin-Bell
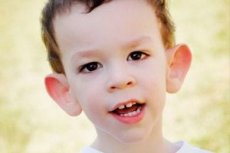
Come riconoscere la malattia nei bambini? Quali sono i primi segni? Nei primi mesi di vita di un bambino, è impossibile riconoscere i sintomi della sindrome di Martin-Bell, se non per una diminuzione del tono muscolare. Dopo un anno, il quadro clinico della malattia è più evidente: il bambino inizia a camminare e parlare tardi, a volte la parola è completamente assente. È iperattivo, agita le braccia in modo casuale, ha paura della folla e del rumore, è testardo, ha forti scoppi d'ira, instabilità emotiva, crisi epilettiche, non stabilisce il contatto visivo. Nei pazienti con sindrome di Martin-Bell, la malattia è rivelata anche dal loro aspetto: orecchie sporgenti e grandi, fronte pesante, viso allungato, mento sporgente, strabismo, mani e piedi larghi. Sono inoltre caratterizzati da disturbi endocrini: spesso sovrappeso, obesità, testicoli grandi negli uomini, pubertà precoce.

Tra i pazienti con sindrome di Martin-Bell, il livello di intelligenza varia notevolmente: da un lieve ritardo mentale a casi gravi. Se una persona normale ha un quoziente intellettivo (QI) medio di 100, e un genio ne ha 130, le persone predisposte alla malattia ne hanno 35-70.
Tutti i sintomi clinici della patologia possono essere caratterizzati da una triade di manifestazioni principali:
- oligofrenia (QI 35-50);
- dismorfofobia (si osservano orecchie a sventola e prognatismo);
- macroorchidismo, che si manifesta dopo l'inizio della pubertà.
Circa l'80% dei pazienti presenta anche prolasso della valvola bicuspide.
Tuttavia, la forma completa della sindrome si manifesta solo nel 60% dei pazienti. Nel 10% dei casi si riscontra solo ritardo mentale e nei restanti la malattia si sviluppa con una diversa combinazione di sintomi.
Tra i primi segni della malattia, che si manifestano in età precoce:
- il bambino malato presenta un ritardo mentale significativo rispetto allo sviluppo dei suoi coetanei;
- disturbi dell'attenzione e della concentrazione;
- forte testardaggine;
- i bambini cominciano a camminare e a parlare piuttosto tardi;
- si osservano iperattività e disturbi dello sviluppo del linguaggio;
- attacchi di rabbia molto forti e incontrollabili;
- può svilupparsi il mutismo, ovvero la completa mancanza di linguaggio nel bambino;
- il bambino soffre di ansia sociale ed è capace di farsi prendere dal panico a causa di rumori forti o altri suoni forti;
- il bambino agita le braccia in modo incontrollabile e caotico;
- si osserva timidezza, il bambino ha paura di trovarsi in luoghi affollati;
- l'emergere di varie idee ossessive, stato emotivo instabile;
- Il bambino potrebbe essere riluttante a stabilire un contatto visivo con le persone.
Negli adulti si osservano i seguenti sintomi della patologia:
- aspetto specifico: viso allungato con fronte pesante, orecchie grandi e sporgenti, mento fortemente sporgente;
- piedi piatti, otite e strabismo;
- la pubertà si verifica abbastanza presto;
- può svilupparsi obesità;
- Molto spesso nella sindrome di Martin-Bell si osservano difetti cardiaci;
- negli uomini si osserva l'ingrossamento dei testicoli;
- le articolazioni delle giunture diventano molto mobili;
- il peso e l'altezza aumentano notevolmente.
Diagnostica Sindrome di Martin-Bell
Per diagnosticare la sindrome di Martin Bell, è necessario contattare un genetista qualificato. La diagnosi viene effettuata dopo specifici test genetici che consentono di identificare il cromosoma difettoso.
Test
In una fase precoce della malattia, viene utilizzato un metodo citogenetico, in cui un frammento di materiale cellulare viene prelevato dal paziente, a cui viene poi aggiunto acido folico per indurre alterazioni cromosomiche. Dopo un certo periodo di tempo, viene identificata un'area del cromosoma in cui si osserva un assottigliamento evidente: questo è un segno della presenza della sindrome dell'X fragile.
Tuttavia, questo test non è adatto per la diagnosi nelle fasi avanzate della malattia, perché la sua accuratezza è ridotta dall'uso diffuso di multivitaminici contenenti acido folico.
La diagnosi integrata della sindrome di Martin-Bell è un esame genetico molecolare, che consiste nella determinazione del numero delle cosiddette ripetizioni trinucleotidiche nel gene.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Diagnostica strumentale
Un metodo altamente specifico di diagnostica strumentale è la PCR (reazione a catena della polimerasi), che consente di studiare la struttura dei residui amminoacidici contenuti nel cromosoma X e di determinare così la presenza della sindrome di Martin Bell.
Esiste anche un metodo diagnostico patologico separato, ancora più specifico: una combinazione di PCR e rilevazione mediante elettroforesi capillare. Questo metodo è altamente accurato e rileva la patologia cromosomica nelle pazienti con insufficienza ovarica primaria e sindrome atassica.
La presenza del difetto può essere determinata dopo aver eseguito la diagnosi tramite EEG. I pazienti affetti da questa patologia presentano un'attività cerebrale bioelettrica simile.
Quali test sono necessari?
Diagnosi differenziale
I metodi differenziati che aiutano a sospettare la sindrome includono:
- clinico - il 97,5% dei pazienti presenta segni evidenti di ritardo mentale (moderato o profondo); il 62% presenta orecchie grandi e sporgenti; il 68,4% presenta mento e fronte grandi e sporgenti; il 68,4% dei ragazzi presenta testicoli ingrossati, il 41,4% presenta peculiarità del linguaggio (velocità di eloquio irregolare, volume incontrollabile, ecc.);
- citogenico: il sangue viene esaminato per la coltura dei linfociti, viene determinato il numero di cellule con cromosoma X fragile ogni 100 cellule studiate;
- Elettroencefalografia: vengono registrate le variazioni degli impulsi elettrici del cervello specifici della sindrome di Martin-Bell.
Chi contattare?
Trattamento Sindrome di Martin-Bell
Nel trattamento dei pazienti adulti, vengono utilizzati antidepressivi con psicostimolanti. Il percorso della terapia farmacologica è costantemente monitorato da uno psicologo e uno psichiatra. Inoltre, presso cliniche private vengono eseguite procedure di microiniezione con farmaci come Cerebrolysin (o suoi derivati), così come citomedine (come Solcoseryl o Lidase).
Nello sviluppo della sindrome atassica, vengono utilizzati farmaci anticoagulanti e nootropi. Inoltre, vengono prescritte miscele di aminoacidi e angioprotettori. Alle donne con insufficienza ovarica primaria viene prescritto un trattamento correttivo a base di erbe medicinali ed estrogeni.
Anche gli antagonisti del recettore della glutammina trovano impiego nella terapia.
Tradizionalmente, il trattamento della sindrome di Martin-Bell prevede l'uso di farmaci che agiscono sui sintomi della malattia, ma non sulla sua causa. Questa terapia prevede la prescrizione di antidepressivi, neurolettici e psicostimolanti. Non tutti i farmaci sono indicati per l'uso nei bambini, quindi l'elenco dei farmaci è piuttosto limitato. I neurolettici che possono essere utilizzati dopo i 3 anni (l'età minima per la loro prescrizione) includono l'aloperidolo in gocce e compresse, la clorpromazina in soluzione e la periciazina in gocce. Pertanto, la dose di aloperidolo per i bambini viene calcolata in base al peso corporeo. Per gli adulti, la dose viene prescritta individualmente. Si assume per via orale, iniziando con 0,5-5 mg 2-3 volte al giorno, per poi aumentare gradualmente fino a 10-15 mg. Al miglioramento, si passa a una dose inferiore per mantenere la condizione raggiunta. In caso di agitazione psicomotoria, vengono prescritti 5-10 mg per via intramuscolare o endovenosa, con possibili ripetizioni dopo 30-40 minuti. La dose giornaliera non deve superare i 100 mg. Sono possibili effetti collaterali come nausea, vomito, spasmi muscolari, aumento della pressione, aritmie, ecc. Gli anziani devono adottare particolari precauzioni, poiché sono stati segnalati casi di arresto cardiaco improvviso e possono verificarsi discinesie tardive (movimenti involontari).
Gli antidepressivi aumentano l'attività delle strutture cerebrali, alleviano la depressione, la tensione e migliorano l'umore. Questi farmaci, raccomandati per l'uso dai 5 agli 8 anni per la sindrome di Martin-Bell, includono clomipromina, sertralina, fluoxetina e fluvoxamina. Pertanto, la fluoxetina viene assunta per via orale durante i pasti 1-2 volte (preferibilmente nella prima metà della giornata), iniziando con 20 mg al giorno, aumentando fino a 80 mg se necessario. Agli anziani non si raccomanda una dose superiore a 60 mg. Il ciclo di trattamento è determinato dal medico, ma non deve durare più di 5 settimane.
Possibili effetti collaterali: vertigini, ansia, tinnito, perdita di appetito, tachicardia, edema, ecc. È richiesta cautela nella prescrizione agli anziani, ai soggetti affetti da malattie cardiovascolari e diabete.
Gli psicostimolanti sono farmaci psicotropi utilizzati per migliorare la percezione degli stimoli esterni: acuiscono l'udito, le reazioni di risposta e la vista.
Il diazepam è prescritto come sedativo per nevrosi, ansia, crisi epilettiche e convulsioni. Viene assunto per via orale, endovenosa, intramuscolare e rettale. Viene prescritto individualmente, a seconda della gravità della malattia, con dosi minime di 5-10 mg e 5-20 mg al giorno. La durata del trattamento è di 2-3 mesi. Per i bambini, la dose viene calcolata tenendo conto del peso corporeo e delle caratteristiche individuali. Gli effetti collaterali includono letargia, apatia, sonnolenza, nausea e stitichezza. È pericoloso assumerlo in concomitanza con l'alcol, poiché può causare dipendenza.
Nel trattamento della sindrome di Martin-Bell, si sono registrati casi di miglioramento della condizione con l'introduzione di farmaci derivati da materiale animale (cervello): cerebrolysate, cerebrolysin, cerebrolysate-M. I componenti principali di questi farmaci sono peptidi che promuovono la produzione di proteine nei neuroni, reintegrando così le proteine mancanti. Cerebrolysin viene somministrato con un getto di 5-10 ml, il ciclo di trattamento consiste in 20-30 iniezioni. Il farmaco viene prescritto ai bambini a partire da un anno di età, somministrato per via intramuscolare ogni giorno a dosi di 1-2 ml per un mese. Sono possibili sedute ripetute di somministrazione. Può causare febbre ed è controindicato alle donne in gravidanza.
Si è tentato di trattare la malattia con acido folico, ma solo l'aspetto comportamentale è migliorato (il livello di aggressività e iperattività è diminuito, il linguaggio è migliorato), mentre a livello intellettivo non è cambiato nulla. Per migliorare le condizioni della malattia, viene prescritto acido folico, sono indicati metodi fisioterapici, logopedia, educazione pedagogica e sociale.
Anche i preparati a base di litio sono considerati efficaci, poiché contribuiscono a migliorare l'adattamento del paziente all'ambiente sociale e l'attività cognitiva. Inoltre, ne regolano anche il comportamento nella società.
L'uso di erbe per la sindrome di Martin-Bell è possibile come antidepressivi. Tra le erbe che aiutano ad alleviare tensione, ansia e migliorare il sonno ci sono valeriana, menta piperita, timo, iperico e camomilla. Gli infusi si preparano come segue: per 1 cucchiaino di erbe secche, è necessario un bicchiere di acqua bollente; i decotti vengono lasciati in infusione per almeno 20 minuti, da assumere principalmente la sera prima di coricarsi o nel pomeriggio. Un cucchiaio di miele sarebbe un'ottima aggiunta.
Trattamento fisioterapico
Per eliminare le manifestazioni neurologiche vengono eseguite speciali procedure fisioterapiche, come esercizi in piscina, rilassamento muscolare e agopuntura.
Trattamento chirurgico
Una fase importante del trattamento è anche considerata la chirurgia plastica, ovvero interventi che aiutano a migliorare l'aspetto del paziente. Vengono eseguiti interventi di chirurgia plastica su arti e padiglioni auricolari, nonché sui genitali. Viene inoltre eseguita la correzione della ginecomastia con epispadia, nonché di altri difetti estetici.
Prevenzione
L'unico metodo per prevenire la malattia è lo screening prenatale delle donne incinte. Esistono esami specifici che consentono la diagnosi precoce della patologia, dopodiché si raccomanda di interrompere la gravidanza. In alternativa, si ricorre alla fecondazione in vitro, che può aiutare il bambino a ereditare un cromosoma X sano.
La prevenzione del paziente dipende dal fatto che la mutazione genetica si sia ripresentata o sia stata ereditata. A tal fine, viene eseguita la diagnosi genetica molecolare. Il fatto che il test non abbia rivelato il "cromosoma X fragile" nei familiari depone a favore della "freschezza" della mutazione, il che significa che il rischio di avere un figlio con la sindrome di Martin-Bell è molto basso. Nelle famiglie in cui sono presenti persone malate, il test contribuirà a evitare casi recidivi.
Previsione
La prognosi della sindrome di Martin-Bell è favorevole per la vita, ma non per la guarigione. L'aspettativa di vita dipende dalla gravità della malattia e dai difetti associati. Il paziente può vivere una vita normale. Nelle forme gravi della sindrome di Martin-Bell, i pazienti sono a rischio di disabilità permanente.
Aspettativa di vita
La sindrome di Martin Bell non ha gravi ripercussioni negative sulla salute, pertanto l'aspettativa di vita della maggior parte delle persone a cui è stata diagnosticata questa patologia non si discosta dagli indicatori standard.