Esperto medico dell'articolo

Nuove pubblicazioni
Prioni - agenti causali delle malattie da prioni
Ultima recensione: 06.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
Le infezioni virali lente sono caratterizzate da criteri particolari:
- un periodo di incubazione insolitamente lungo (mesi, anni);
- una lesione specifica di organi e tessuti, principalmente del sistema nervoso centrale;
- progressione lenta e costante della malattia;
- inevitabile esito fatale.

Alcuni patogeni che causano infezioni virali acute possono anche causare infezioni virali lente. Ad esempio, il virus del morbillo a volte causa la PESS (sclerosi multipla ereditaria), mentre il virus della rosolia causa la rosolia congenita progressiva e la panencefalite da rosolia.
Una tipica infezione virale lenta negli animali è causata dal virus visna/madi, un retrovirus. È l'agente eziologico dell'infezione virale lenta e della polmonite progressiva negli ovini. La sostanza bianca del cervello viene distrutta, si sviluppa paralisi (visna - deperimento); si verifica un'infiammazione cronica di polmoni e milza.
Malattie simili nelle loro caratteristiche alle infezioni virali lente sono causate dai prioni, gli agenti causali delle infezioni da prioni. Le malattie da prioni sono un gruppo di disturbi progressivi del sistema nervoso centrale di esseri umani e animali. Negli esseri umani, la funzionalità del sistema nervoso centrale è compromessa, si verificano cambiamenti di personalità e disturbi del movimento. I sintomi della malattia durano solitamente da diversi mesi a diversi anni, terminando con la morte. In precedenza, le infezioni da prioni venivano considerate insieme ai cosiddetti agenti causali delle infezioni virali lente.
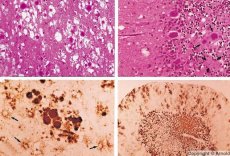
Alcuni agenti che causano le malattie da prioni si accumulano inizialmente nei tessuti linfoidi. I prioni, penetrando nel cervello, si accumulano in grandi quantità, causando amiloidosi (disproteinosi extracellulare, caratterizzata dalla deposizione di amiloide con sviluppo di atrofia e sclerosi del tessuto) e astrocitosi (proliferazione della neuroglia astrocitaria, iperproduzione di fibre gliali). Si formano fibrille, aggregati proteici o amiloidi e alterazioni spongiformi nel cervello (encefalopatie spongiformi trasmissibili). Di conseguenza, si verificano alterazioni del comportamento, compromissione della coordinazione dei movimenti, esaurimento con esito fatale. Non si forma immunità. Le malattie da prioni sono malattie conformazionali che si sviluppano a causa di un errato ripiegamento (violazione della corretta conformazione) delle proteine cellulari necessarie per il normale funzionamento dell'organismo. Le vie di trasmissione dei prioni sono molteplici:
- via alimentare - prodotti infetti di origine animale, additivi alimentari derivati da organi bovini crudi, ecc.:
- trasmissione tramite trasfusione di sangue, somministrazione di farmaci di origine animale, trapianto di organi e tessuti, utilizzo di strumenti chirurgici e odontoiatrici infetti;
- trasmissione attraverso preparati immunobiologici (è nota l'infezione di 1500 pecore con PrP''' mediante vaccino contro il formolo cerebrale proveniente da pecore malate).
I prioni patologici, una volta penetrati nell'intestino, vengono trasportati nel sangue e nella linfa. Dopo la replicazione periferica nella milza, nell'appendice, nelle tonsille e in altri tessuti linfoidi, vengono trasferiti al cervello attraverso i nervi periferici (neuroinvasione). È possibile la penetrazione diretta dei prioni nel cervello attraverso la barriera emato-encefalica. In precedenza, si riteneva che il sistema nervoso centrale fosse l'unico tessuto in cui si accumulassero i prioni patologici, ma sono comparsi studi che hanno modificato questa ipotesi. Si è scoperto che l'accumulo di prioni nella milza è associato all'aumento e al funzionamento delle cellule dendritiche follicolari.
 [
[ Proprietà dei prioni
L'isoforma cellulare normale della proteina prionica con un peso molecolare di 33-35 kDa è determinata dal gene della proteina prionica (il gene prione - PrNP è localizzato sul 20° cromosoma umano). Il gene normale appare sulla superficie cellulare (ancorato alla membrana dalla glicoproteina della molecola), sensibile alla proteasi. Regola la trasmissione degli impulsi nervosi, i cicli giornalieri, i processi di ossidazione, partecipa al metabolismo del rame nel sistema nervoso centrale e alla regolazione della divisione delle cellule staminali del midollo osseo. Inoltre, il gene prione si trova nella milza, nei linfonodi, nella pelle, nel tratto gastrointestinale e nelle cellule dendritiche follicolari.
Proliferazione di prioni patologici
La trasformazione dei prioni in forme alterate avviene quando l'equilibrio cineticamente controllato tra di essi viene interrotto. Il processo è favorito da un aumento della quantità di prione patologico (PrP) o esogeno. PrP è una proteina normale ancorata alla membrana cellulare. PrP' è una proteina globulare idrofobica che forma aggregati con se stessa e PrP'' sulla superficie cellulare: di conseguenza, PrP' si trasforma in PrP'' e il ciclo continua. La forma patologica di PrP''' si accumula nei neuroni, conferendo alla cellula un aspetto spugnoso.
Kuru
Malattia da prioni, un tempo comune tra i papuani (il cui significato è "tremore o scuotimento") nella parte orientale dell'isola di Nuova Guinea. Le proprietà infettive della malattia sono state dimostrate da K. Gajdusek. Il patogeno si trasmette attraverso il cibo a seguito di cannibalismo rituale, ovvero mangiando il cervello infetto da prioni e non cotto a sufficienza di parenti defunti. A causa del danno al sistema nervoso centrale, i movimenti e l'andatura sono compromessi, compaiono brividi ed euforia ("morte che ride"). Il periodo di incubazione dura dai 5 ai 30 anni. Il paziente muore dopo un anno.
Malattia di Creutzfeldt-Jakob
La malattia da prioni si manifesta con demenza, disturbi visivi e cerebellari e disturbi del movimento, con esito fatale dopo 4-5 mesi di malattia nella variante classica della malattia di Creutzfeldt-Jakob e dopo 3-14 mesi nella nuova variante della malattia di Creutzfeldt-Jakob. Il periodo di incubazione può raggiungere i 20 anni. Sono possibili diverse vie di infezione e cause della malattia:
- quando si consumano prodotti animali non trattati termicamente in modo sufficiente, come carne e cervello di mucche affette da encefalopatia spongiforme bovina;
- durante il trapianto di tessuti, come il trapianto di cornea, la trasfusione di sangue, l'uso di ormoni e altre sostanze biologicamente attive di origine animale, l'uso di catgut, strumenti chirurgici contaminati o non sufficientemente sterilizzati, manipolazioni prosettoriali;
- in caso di iperproduzione di PrR e altre condizioni che stimolano il processo di conversione di PrR' in PrR".
La malattia può anche svilupparsi a seguito di una mutazione o di un'inserzione nella regione del gene prionico. La natura familiare della malattia è comune a causa della predisposizione genetica alla malattia di Creutzfeldt-Jakob. Nella nuova variante della malattia di Creutzfeldt-Jakob, i disturbi si sviluppano in età più giovane (età media 28 anni), a differenza della variante classica (età media 65 anni). Nella nuova variante della malattia di Creutzfeldt-Jakob, la proteina prionica anomala si accumula non solo nel sistema nervoso centrale, ma anche nei tessuti linforeticolari, comprese le tonsille.
Sindrome di Gerstmann-Sträussler-Scheinker
Malattia da prioni ereditaria, accompagnata da demenza, ipotonia, disturbi della deglutizione (disfagia) e disartria. Spesso ha natura familiare. Il periodo di incubazione è compreso tra 5 e 30 anni. La malattia si manifesta tra i 50 e i 60 anni e la sua durata varia dai 5 ai 13 anni.
Insonnia ereditaria fatale
Malattia autoimmune con insonnia progressiva, iperreattività simpatica (ipertensione, ipertermia, iperidrosi, tachicardia), tremori, atassia, multicloni, allucinazioni. Il sonno è gravemente disturbato. La morte sopraggiunge con progressione dell'insufficienza cardiovascolare.
Raschiare
La scrapie (dall'inglese scrape - raschiare) è una malattia da prioni che colpisce pecore e capre (scabbia), che si manifesta con danni al sistema nervoso centrale, disturbi progressivi del movimento, forte prurito cutaneo (scabbia) e termina con la morte dell'animale.
Encefalopatia spongiforme bovina
Malattia dei bovini caratterizzata da danni al sistema nervoso centrale, compromissione della coordinazione dei movimenti e morte inevitabile dell'animale. L'epidemia della malattia scoppiò per la prima volta in Gran Bretagna. Fu associata all'alimentazione degli animali con farine di carne e ossa contenenti prioni patologici. Il periodo di incubazione varia da 1,5 a 15 anni. Il cervello, il midollo spinale e i bulbi oculari degli animali sono le zone più colpite.
Diagnostica di laboratorio delle malattie da prioni
Durante la diagnosi, si notano alterazioni spongiformi nel cervello, astrocitosi (gliosi) e assenza di infiltrati infiammatori. Il cervello viene colorato per la presenza di amiloide. Marcatori proteici delle patologie cerebrali da prioni vengono rilevati nel liquido cerebrospinale (mediante ELISA). Viene eseguita l'analisi genetica del gene del prione (PCR).
Prevenzione delle malattie da prioni
Per la decontaminazione di strumenti e oggetti ambientali si raccomanda l'autoclavaggio (a 134 °C per 18 min; a 121 °C per 1 h), l'incenerimento e un trattamento aggiuntivo con candeggina e una soluzione di NaCl a un valore normale per 1 h. Per la profilassi non specifica, sono state introdotte restrizioni all'uso di medicinali di origine animale ed è vietata la produzione di ormoni ipofisari di origine animale. Il trapianto di dura madre è limitato. Si utilizzano guanti di gomma quando si maneggiano i fluidi dialogici dei pazienti.