Esperto medico dell'articolo

Nuove pubblicazioni
Malattia di Charcot-Marie-Tooth.
Ultima recensione: 12.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
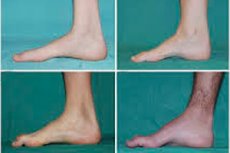
L'atrofia muscolare peronea, sindrome o malattia di Charcot-Marie-Tooth, è un gruppo di malattie ereditarie croniche con danni ai nervi periferici.
Secondo l'ICD-10, nella sezione dedicata alle malattie del sistema nervoso, il codice per questa malattia è G60.0 (neuropatia motoria e sensoriale ereditaria). È inclusa anche nell'elenco delle malattie orfane.
Epidemiologia
Secondo le statistiche cliniche, la prevalenza di tutti i tipi della malattia di Charcot-Marie-Tooth è di 19 casi ogni 100 mila abitanti (secondo altre fonti, un caso ogni 2,5-10 mila abitanti).
La CMT di tipo 1 rappresenta circa due terzi dei casi (un caso ogni 5.000-7.000 abitanti) e quasi il 70% di essi è associato alla duplicazione del gene PMP22. Oltre 1,2 milioni di persone in tutto il mondo soffrono di questo tipo di malattia.
L'incidenza della CMT di tipo 4 è stimata in 1-5 casi ogni 10.000 bambini. [ 1 ]
Le cause Malattia di Charcot-Marie-Tooth
Secondo la classificazione delle sindromi polineuropatiche, l'atrofia muscolare peronea (fibulare), l'amiotrofia neurale di Charcot-Marie-Tooth o la malattia di Charcot-Marie-Tooth (abbreviata in CMT) si riferiscono a polineuropatie motorie-sensoriali geneticamente determinate. [ 2 ]
Ciò significa che le cause della sua comparsa sono mutazioni genetiche. E a seconda della natura delle deviazioni genetiche, si distinguono i principali tipi o generi di questa sindrome: demielinizzante e assonale. Il primo gruppo include la malattia di Charcot-Marie-Tooth di tipo 1 (CMT1), che si verifica a causa della duplicazione del gene PMP22 sul cromosoma 17, che codifica per la proteina mielinica periferica transmembrana 22. Di conseguenza, si verificano demielinizzazione segmentale della guaina assonale (processi delle cellule nervose) e una riduzione della velocità di conduzione del segnale nervoso. Inoltre, possono essere presenti mutazioni in altri geni.
La forma assonale è la malattia di Charcot-Marie-Tooth di tipo 2 (CMT2), che colpisce gli assoni stessi ed è associata ad alterazioni patologiche del gene MFN2, localizzato nel locus 1p36.22, che codifica per la proteina di membrana mitofusina-2, necessaria per la fusione dei mitocondri e la formazione di reti mitocondriali funzionali all'interno delle cellule nervose periferiche. Esistono più di una dozzina di sottotipi di CMT2 (con mutazioni in geni specifici).
È importante sottolineare che attualmente sono stati identificati più di cento geni il cui danno, trasmesso per via ereditaria, causa vari sottotipi di malattia di Charcot-Marie-Tooth. Ad esempio, mutazioni nel gene RAB7 portano alla CMT di tipo 2B; l'alterazione del gene SH3TC2 (che codifica per una delle proteine di membrana delle cellule di Schwann) causa la CMT di tipo 4C, che si manifesta nell'infanzia ed è caratterizzata da demielinizzazione dei neuroni motori e sensoriali (esistono una dozzina e mezza di forme di tipo 4 di questa malattia).
Una rara forma di CMT di tipo 3 (detta sindrome di Dejerine-Sottas) inizia a svilupparsi nella prima infanzia ed è causata da mutazioni nei geni PMP22, MPZ, EGR2 e altri.
Quando la CMT di tipo 5 si manifesta tra i 5 e i 12 anni, si osserva non solo neuropatia motoria (sotto forma di paraparesi spastica degli arti inferiori), ma anche danni ai nervi ottici e uditivi.
Debolezza muscolare e atrofia ottica (con perdita della vista), nonché problemi di equilibrio, sono tipici della CMT di tipo 6. Nella malattia di Charcot-Marie-Tooth di tipo 7 non è presente solo una neuropatia motoria-sensoriale, ma anche una malattia della retina sotto forma di retinite pigmentosa.
Più comune tra i maschi, la CMT legata all'X o malattia di Charcot-Marie-Tooth con tetraparesi degli arti (debolezza del movimento di braccia e gambe) è un tipo demielinizzante e si pensa che derivi da una mutazione nel gene GJB1 sul braccio lungo del cromosoma X, che codifica la connessina 32, una proteina transmembrana delle cellule di Schwann e degli oligodendrociti che regola la trasmissione dei segnali nervosi. [ 3 ]
Fattori di rischio
Il principale fattore di rischio per la CMT è la storia familiare della malattia, cioè nei parenti stretti.
Secondo i genetisti, se entrambi i genitori sono portatori del gene autosomico recessivo per la malattia di Charcot-Marie-Tooth, il rischio di avere un figlio che sviluppi questa malattia è del 25%. Il rischio che il figlio sia portatore sano di questo gene (ma non presenti alcun sintomo) è stimato al 50%.
In caso di ereditarietà legata al cromosoma X (quando il gene mutato è presente sul cromosoma X della donna), c'è un rischio del 50% che la madre trasmetta il gene al figlio, che svilupperà la CMT. La malattia potrebbe non manifestarsi alla nascita di una figlia femmina, ma i figli maschi (nipoti) della figlia potrebbero ereditare il gene difettoso e la malattia si svilupperà.
Patogenesi
In qualsiasi tipo di malattia di Charcot-Marie-Tooth, la sua patogenesi è causata da un'anomalia ereditaria dei nervi periferici: motori (di movimento) e sensoriali (sensibili).
Se la CMT è di tipo demielinizzante, la distruzione o il difetto della guaina mielinica che protegge gli assoni dei nervi periferici provoca un rallentamento nella trasmissione degli impulsi nervosi nel sistema nervoso periferico, tra cervello, muscoli e organi sensoriali.
Nella forma assonale della malattia, vengono colpiti gli assoni stessi, il che influisce negativamente sulla forza dei segnali nervosi, che non è sufficiente per la piena stimolazione dei muscoli e degli organi sensoriali.
Leggi anche:
Come si trasmette la sindrome di Charcot-Marie-Tooth? I geni difettosi possono essere ereditati con modalità autosomica dominante o autosomica recessiva.
Il tipo più comune, l'ereditarietà autosomica dominante, si verifica quando è presente una sola copia del gene mutato (portata da uno dei genitori). E la probabilità di trasmettere la CMT a ogni bambino nato è stimata al 50%. [ 4 ]
Nell'ereditarietà autosomica recessiva, sono necessarie due copie del gene difettoso (una da ciascun genitore che non mostra alcun segno della malattia) affinché si sviluppi la malattia.
Nel 40-50% dei casi si verifica una demielinizzazione ereditaria autosomica dominante, cioè CMT di tipo 1; nel 12-26% dei casi, CMT assonale, cioè di tipo 2. E nel 10-15% dei casi si osserva un'ereditarietà legata al cromosoma X. [ 5 ]
Sintomi Malattia di Charcot-Marie-Tooth
In genere, i primi segni di questa malattia iniziano a manifestarsi nell'infanzia e nell'adolescenza e si sviluppano gradualmente nel corso della vita, sebbene la sindrome possa manifestarsi anche in seguito. La combinazione dei sintomi è variabile e la velocità di progressione della malattia, così come la sua gravità, è impossibile da prevedere.
I sintomi tipici della fase iniziale includono un aumento della stanchezza generale; una diminuzione del tono (debolezza) dei muscoli di piedi, caviglie e stinchi; mancanza di riflessi. Ciò rende difficile il movimento del piede e porta a disbasia (disturbo dell'andatura) sotto forma di un maggiore sollevamento delle gambe, spesso con frequenti inciampi e cadute. I segni della malattia di Charcot-Marie-Tooth in un bambino piccolo possono includere una marcata goffaggine e difficoltà di deambulazione insolite per l'età associate a piede cadente bilaterale. Sono caratteristiche anche le deformità del piede: arco plantare elevato (piede cavo) o piedi piatti gravi, dita curve (a forma di martello).
Nel caso in cui il bambino cammini sulle punte in presenza di ipotonia muscolare, un neurologo può sospettare che sia affetto da CMT di tipo 4, che può portare i bambini ad essere incapaci di camminare entro l'adolescenza.
Con il progredire della malattia, l'atrofia muscolare e la debolezza si diffondono agli arti superiori, rendendo difficile l'esecuzione di movimenti fini e l'esecuzione di normali compiti manuali. La riduzione della sensibilità tattile e della capacità di percepire il caldo e il freddo, nonché l'intorpidimento di piedi e mani, indicano un danno agli assoni dei nervi sensoriali.
Nella malattia di Charcot-Marie-Tooth di tipo 3 e 6 che si manifesta nell'infanzia, si osservano atassia sensoriale (compromissione della coordinazione dei movimenti e dell'equilibrio), spasmi muscolari e tremori, danni al nervo facciale, atrofia del nervo ottico con nistagmo e perdita dell'udito.
Nelle fasi avanzate possono manifestarsi tremori incontrollabili e frequenti crampi muscolari; problemi di movimento possono portare allo sviluppo di dolori: muscolari, articolari, neuropatici.
Complicazioni e conseguenze
La malattia di Charcot-Marie-Tooth può avere complicazioni e conseguenze come:
- distorsioni e fratture più frequenti;
- contratture associate all'accorciamento dei muscoli e dei tendini periarticolari;
- scoliosi (curvatura della colonna vertebrale);
- problemi respiratori – quando le fibre nervose che innervano i muscoli del diaframma sono danneggiate:
- perdita della capacità di muoversi in modo autonomo.
Diagnostica Malattia di Charcot-Marie-Tooth
La diagnosi comprende l'esame clinico, l'anamnesi (inclusa la storia familiare), l'esame neurologico e sistemico.
Vengono eseguiti test per verificare l'ampiezza del movimento, la sensibilità e i riflessi tendinei. La conduzione nervosa può essere valutata utilizzando la diagnostica strumentale: elettromiografia o elettroneuromiografia. Potrebbero essere necessari anche ultrasuoni o risonanza magnetica. [ 6 ]
I test genetici o del DNA per rilevare le mutazioni genetiche più comuni che causano la CMT in un campione di sangue sono limitati perché i test del DNA non sono attualmente disponibili per tutti i tipi di malattia. Per ulteriori informazioni, consultare la sezione Test genetici.
In alcuni casi viene eseguita una biopsia del nervo periferico (solitamente il nervo surale).
Diagnosi differenziale
La diagnosi differenziale comprende altre neuropatie periferiche, la distrofia muscolare di Duchenne, le sindromi mielopatiche e miasteniche, la neuropatia diabetica, le mielopatie nella sclerosi laterale amiotrofica e multipla, la sindrome di Guillain-Barré, i traumi al nervo peroneo e la sua atrofia (anche quando schiacciato tra i dischi lombari della colonna vertebrale), i danni al cervelletto o al talamo, nonché gli effetti collaterali della chemioterapia (durante il trattamento con citostatici come la vincristina o il paclitaxel). [ 7 ]
Chi contattare?
Trattamento Malattia di Charcot-Marie-Tooth
Oggi, il trattamento per questa malattia ereditaria comprende la terapia fisica (finalizzata al rafforzamento e all'allungamento dei muscoli); la terapia occupazionale (che aiuta i pazienti con debolezza muscolare alle mani); e l'uso di dispositivi ortopedici per facilitare la deambulazione. Se necessario, vengono prescritti antidolorifici o anticonvulsivanti. [ 8 ]
Nei casi di piede piatto grave si può effettuare l'osteotomia, mentre in caso di deformazione del tallone è indicata la correzione chirurgica: l'artrodesi. [ 9 ]
Sono in corso ricerche sia sulla componente genetica della malattia che sui suoi metodi di trattamento. L'uso di cellule staminali, di alcuni ormoni, della lecitina o dell'acido ascorbico non ha ancora prodotto risultati positivi.
Ma grazie a recenti ricerche, qualcosa di nuovo potrebbe effettivamente emergere nel trattamento della malattia di Charcot-Marie-Tooth nel prossimo futuro. Pertanto, dal 2014, l'azienda francese Pharnext ha sviluppato e, dalla metà del 2019, sono in corso studi clinici per il farmaco PXT3003 per il trattamento della CMT di tipo 1 negli adulti, che sopprime l'espressione aumentata del gene PMP22, migliora la mielinizzazione dei nervi periferici e allevia i sintomi neuromuscolari.
Gli specialisti dell'azienda farmaceutica Sarepta Therapeutics (USA) stanno lavorando alla creazione di una terapia genica per la malattia di Charcot-Marie-Tooth di tipo 1. Questa terapia utilizzerà un virus adeno-associato (AAV) innocuo del genere Dependovirus con un genoma di DNA lineare a singolo filamento, che trasferirà nell'organismo il gene NTF3, che codifica per la proteina neurotrofina-3 (NT-3), necessaria per il funzionamento delle cellule nervose di Schwann.
Helixmith inizierà le sperimentazioni cliniche della terapia genica sudcoreana Engensis (VM202) entro la fine del 2020 per trattare i sintomi muscolari nella CMT di tipo 1. [ 10 ]
Prevenzione
La prevenzione della CMT può essere effettuata tramite consulenza genetica per i futuri genitori, soprattutto se un membro della coppia ha una storia familiare della malattia. Tuttavia, sono stati identificati casi di mutazioni geniche puntiformi de novo, ovvero in assenza della malattia nella storia familiare.
Durante la gravidanza, la probabilità che il nascituro sia affetto dalla malattia di Charcot-Marie-Tooth può essere verificata mediante una biopsia dei villi coriali (dalla 10a alla 13a settimana di gestazione) e un'analisi del liquido amniotico (tra la 15a e la 18a settimana).
Previsione
In generale, la prognosi per i diversi tipi di malattia di Charcot-Marie-Tooth dipende dalla gravità clinica, ma in tutti i casi la malattia progredisce lentamente. Molti pazienti presentano disabilità, sebbene ciò non riduca l'aspettativa di vita.