Esperto medico dell'articolo

Nuove pubblicazioni
Nefrite ereditaria (sindrome di Alport) nei bambini
Ultima recensione: 05.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
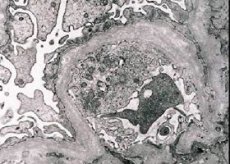
La nefrite ereditaria (sindrome di Alport) è una glomerulopatia ereditaria non immune di origine genetica, che si manifesta con ematuria (talvolta associata a proteinuria), progressivo declino della funzionalità renale con sviluppo di insufficienza renale cronica, spesso associata a sordità neurosensoriale e deficit visivo.
La malattia fu descritta per la prima volta nel 1902 da LG Guthrie, che osservò una famiglia in cui l'ematuria era presente in diverse generazioni. Nel 1915, AF Hurst descrisse lo sviluppo di uremia in membri della stessa famiglia. Nel 1927, A. Alport identificò per la prima volta la perdita dell'udito in diversi parenti affetti da ematuria. Negli anni '50, furono descritte lesioni oculari in una malattia simile. Nel 1972, in pazienti con ematuria ereditaria, durante uno studio morfologico del tessuto renale, Hinglais et al. rivelarono un'espansione e una stratificazione irregolari delle membrane basali glomerulari. Nel 1985, fu identificata la base genetica della nefrite ereditaria: una mutazione nel gene del collagene di tipo IV (Fiengold et al., 1985).
Lo studio della natura genetica della malattia ci ha permesso di concludere che le differenze nelle manifestazioni fenotipiche della nefrite ereditaria (con o senza perdita dell'udito) sono dovute al grado di espressione del gene mutante. Pertanto, attualmente, tutte le varianti cliniche sono considerate manifestazioni di un'unica malattia e il termine "nefrite ereditaria" è sinonimo di "sindrome di Alport".
Secondo studi epidemiologici, la nefrite ereditaria si verifica con una frequenza di 17 casi ogni 100.000 bambini.
 [
[ Cause della sindrome di Alport
La base genetica della malattia è una mutazione nel gene della catena α-5 del collagene di tipo IV. Questo tipo è universale per le membrane basali del rene, dell'apparato cocleare, della capsula del cristallino, della retina e della cornea dell'occhio, come dimostrato da studi che utilizzano anticorpi monoclonali contro questa frazione di collagene. Recentemente, è stata indicata la possibilità di utilizzare sonde di DNA per la diagnosi prenatale della nefrite ereditaria.
Viene sottolineata l'importanza di testare tutti i membri della famiglia con sonde di DNA per identificare i portatori del gene mutante, il che è di grande importanza per condurre una consulenza medica e genetica alle famiglie con questa malattia. Tuttavia, fino al 20% delle famiglie non ha parenti affetti da malattie renali, il che suggerisce un'alta frequenza di mutazioni spontanee del gene anomalo. La maggior parte dei pazienti con nefrite ereditaria ha in famiglia individui con malattie renali, perdita dell'udito e patologie della vista; i matrimoni consanguinei tra persone con uno o più antenati sono importanti, poiché nel matrimonio tra individui imparentati aumenta la probabilità di ricevere gli stessi geni da entrambi i genitori. Sono state identificate vie di trasmissione autosomica dominante, autosomica recessiva e dominante legata al cromosoma X.
Nei bambini si distinguono più comunemente tre tipi di nefrite ereditaria: la sindrome di Alport, la nefrite ereditaria senza perdita dell'udito e l'ematuria benigna familiare.
La sindrome di Alport è una nefrite ereditaria con deficit uditivo. È causata da un difetto combinato nella struttura del collagene della membrana basale glomerulare di reni, orecchie e occhi. Il gene della sindrome di Alport classica è localizzato nel locus 21-22 q del braccio lungo del cromosoma X. Nella maggior parte dei casi, è ereditato in modo dominante, legato al cromosoma X. A questo proposito, la sindrome di Alport è più grave negli uomini, poiché nelle donne la funzione del gene mutante è compensata da un allele sano del secondo cromosoma, non danneggiato.
La base genetica per lo sviluppo della nefrite ereditaria è rappresentata da mutazioni nei geni delle catene alfa del collagene di tipo IV. Sono note sei catene alfa del collagene di tipo IV G: i geni delle catene a5 e a6 (Col4A5 e Col4A5) sono localizzati sul braccio lungo del cromosoma X, nella zona 21-22q; i geni delle catene a3 e a4 (Col4A3 e Col4A4) sono localizzati sul secondo cromosoma; i geni delle catene a1 e a2 (Col4A1 e Col4A2) sono localizzati sul tredicesimo cromosoma.
Nella maggior parte dei casi (80-85%), viene rilevata una trasmissione ereditaria legata al cromosoma X della malattia, associata a danni al gene Col4A5 dovuti a delezione, mutazioni puntiformi o anomalie dello splicing. Attualmente sono state identificate più di 200 mutazioni del gene Col4A5, responsabili dell'interruzione della sintesi delle catene α5 del collagene di tipo IV. Con questo tipo di trasmissione ereditaria, la malattia si manifesta nei bambini di entrambi i sessi, ma nei maschi è più grave.
Le mutazioni nei loci dei geni Col4A3 e Col4A4, responsabili della sintesi delle catene a3 e a4 del collagene di tipo IV, sono ereditarie con modalità autosomica. Secondo la ricerca, la trasmissione autosomica dominante si osserva nel 16% dei casi di nefrite ereditaria, mentre quella recessiva nel 6% dei pazienti. Sono note circa 10 varianti di mutazioni dei geni Col4A3 e Col4A4.
Il risultato delle mutazioni è una violazione dei processi di assemblaggio del collagene di tipo IV, che porta a una compromissione della sua struttura. Il collagene di tipo IV è uno dei componenti principali della membrana basale glomerulare, dell'apparato cocleare e del cristallino dell'occhio, la cui patologia verrà rilevata nella clinica della nefrite ereditaria.
Il collagene di tipo IV, che fa parte della membrana basale glomerulare, è costituito principalmente da due catene α1 (IV) e una catena α2 (IV), e contiene anche catene α3, α4 e α5. Nella maggior parte dei casi, nell'ereditarietà legata al cromosoma X, la mutazione del gene Col4A5 è accompagnata dall'assenza delle catene α3, α4, α5 e α6 nella struttura del collagene di tipo IV, e da un aumento del numero di catene α1 e α2 nella membrana basale glomerulare. Il meccanismo di questo fenomeno non è chiaro; si presume che la causa siano cambiamenti post-trascrizionali nell'mRNA.
L'assenza delle catene a3, a4 e a5 nella struttura del collagene di tipo IV delle membrane basali glomerulari ne determina l'assottigliamento e la fragilità nelle fasi precoci della sindrome di Alport, che si manifesta clinicamente più spesso con ematuria (meno frequentemente con ematuria associata a proteinuria o solo proteinuria), perdita dell'udito e lenticono. L'ulteriore progressione della malattia porta all'ispessimento e alla ridotta permeabilità delle membrane basali nelle fasi avanzate della malattia, con proliferazione dei collageni di tipo V e VI, che si manifesta con un aumento della proteinuria e una riduzione della funzionalità renale.
La natura della mutazione alla base della nefrite ereditaria determina in larga misura la sua manifestazione fenotipica. In caso di delezione del cromosoma X con mutazione simultanea dei geni Col4A5 e Col4A6, responsabili della sintesi delle catene α5 e α6 del collagene di tipo IV, la sindrome di Alport si associa a leiomiomatosi dell'esofago e dei genitali. Secondo i dati della ricerca, in caso di mutazione del gene Col4A5 associata a delezione, si osserva una maggiore gravità del processo patologico, una combinazione di danno renale con manifestazioni extrarenali e sviluppo precoce di insufficienza renale cronica, rispetto a una mutazione puntiforme di questo gene.
Morfologicamente, la microscopia elettronica rivela un assottigliamento e una stratificazione delle membrane basali glomerulari (in particolare della lamina densa) e la presenza di granuli elettrondensi. Le lesioni glomerulari possono essere eterogenee nello stesso paziente, da lesioni mesangiali focali minime alla glomerulosclerosi. La glomerulite nella sindrome di Alport è sempre immunonegativa, il che la distingue dalla glomerulonefrite. Le caratteristiche includono lo sviluppo di atrofia tubulare, infiltrazione linfoistiocitaria e la presenza di "cellule schiumose" con inclusioni lipidiche - lipofagi. Con la progressione della malattia, si rivelano un ispessimento e una marcata distruzione delle membrane basali glomerulari.
Vengono rivelate alcune alterazioni del sistema immunitario. I pazienti con nefrite ereditaria presentano un livello ridotto di IgA e una tendenza ad aumentare la concentrazione di IgM nel sangue; il livello di IgG può aumentare nelle fasi iniziali della malattia e diminuire nelle fasi avanzate. Forse, l'aumento della concentrazione di IgM e G è una sorta di reazione compensatoria in risposta alla carenza di IgA.
L'attività funzionale del sistema dei linfociti T è ridotta; si nota una diminuzione selettiva dei linfociti B responsabili della sintesi di Ig A, il legame fagocitico dell'immunità è interrotto, principalmente a causa dell'interruzione della chemiotassi e dei processi di digestione intracellulare nei neutrofili.
Esaminando una biopsia renale in pazienti con sindrome di Alport, i dati di microscopia elettronica rivelano alterazioni ultrastrutturali della membrana basale glomerulare: assottigliamento, alterazione della struttura e frammentazione delle membrane basali glomerulari, con alterazione dello spessore e irregolarità dei contorni. Nelle fasi precoci della nefrite ereditaria, il difetto determina l'assottigliamento e la fragilità delle membrane basali glomerulari.
L'assottigliamento delle membrane glomerulari è un segno più favorevole ed è più comune nelle ragazze. Un segno microscopico elettronico più costante nella nefrite ereditaria è la scissione della membrana basale, e la gravità della sua distruzione è correlata alla gravità del processo.
Sintomi della sindrome di Alport nei bambini
I primi sintomi della sindrome di Alport, sotto forma di sindrome urinaria isolata, vengono rilevati più spesso nei bambini entro i primi tre anni di vita. Nella maggior parte dei casi, la malattia viene diagnosticata casualmente. La sindrome urinaria viene diagnosticata durante una visita preventiva del bambino, prima del ricovero in un asilo nido o durante l'ARVI. In caso di patologia urinaria durante l'ARVI. Nella nefrite ereditaria, a differenza della glomerulonefrite acquisita, non esiste un periodo di latenza.
Nella fase iniziale della malattia, la salute del bambino ne risente poco; una caratteristica è la persistenza e la resistenza della sindrome urinaria. Uno dei segni principali è l'ematuria di varia gravità, osservata nel 100% dei casi. Un aumento del grado di ematuria si osserva durante o dopo infezioni respiratorie, attività fisica o vaccinazioni preventive. La proteinuria nella maggior parte dei casi non supera 1 g/die; all'inizio della malattia può essere incostante, ma con il progredire della malattia aumenta. Periodicamente, nel sedimento urinario può essere presente leucocituria con predominanza di linfociti, associata allo sviluppo di alterazioni interstiziali.
Successivamente, la funzionalità renale parziale è compromessa, le condizioni generali del paziente peggiorano: compaiono intossicazione, debolezza muscolare, ipotensione arteriosa, spesso deficit uditivo (soprattutto nei ragazzi) e talvolta deficit visivo. L'intossicazione si manifesta con pallore, affaticamento e mal di testa. Nella fase iniziale della malattia, la perdita dell'udito è nella maggior parte dei casi rilevata solo tramite audiografia. La perdita dell'udito nella sindrome di Alport può verificarsi in diversi periodi dell'infanzia, ma il più delle volte viene diagnosticata all'età di 6-10 anni. La perdita dell'udito nei bambini inizia con le alte frequenze, raggiungendo un grado significativo nella conduzione aerea e ossea, passando dalla perdita dell'udito conduttore a quella percettivo. La perdita dell'udito può essere uno dei primi sintomi della malattia e può precedere la sindrome urinaria.
Nel 20% dei casi, i pazienti con sindrome di Alport presentano alterazioni degli organi visivi. Le anomalie più frequentemente rilevate sono quelle del cristallino: sferofochia, lenticono anteriore, posteriore o misto e varie forme di cataratta. Nelle famiglie con sindrome di Alport, la miopia è una frequenza significativa. Diversi ricercatori notano costantemente alterazioni perimaculari bilaterali in queste famiglie, sotto forma di granulazioni biancastre o giallastre brillanti nel corpo luteo. Considerano questo segno un sintomo costante ad alto valore diagnostico nella sindrome di Alport. KS Chugh et al. (1993) in uno studio oftalmologico hanno riscontrato nei pazienti con sindrome di Alport una diminuzione dell'acuità visiva nel 66,7% dei casi, lenticono anteriore nel 37,8%, macchie retiniche nel 22,2%, cataratta nel 20% e cheratocono nel 6,7%.
In alcuni bambini con nefrite ereditaria, soprattutto quando si sviluppa insufficienza renale, si osserva un ritardo significativo nello sviluppo fisico. Con il progredire dell'insufficienza renale, si sviluppa ipertensione arteriosa. Nei bambini, viene rilevata più spesso durante l'adolescenza e nelle fasce d'età più avanzate.
I pazienti con nefrite ereditaria sono caratterizzati dalla presenza di vari (più di 5-7) stigmi di dismorfogenesi del tessuto connettivo. Tra gli stigmi del tessuto connettivo nei pazienti, i più comuni sono l'ipertelorismo degli occhi, il palato alto, le anomalie del morso, la forma anomala dei padiglioni auricolari, la curvatura del mignolo delle mani e la "fessura a sandalo" dei piedi. La nefrite ereditaria è caratterizzata dall'uniformità degli stigmi di dismorfogenesi all'interno di una famiglia, nonché da un'elevata frequenza della loro distribuzione tra i parenti dei probandi lungo la cui linea di trasmissione della malattia.
Nelle fasi precoci della malattia, si riscontra una riduzione isolata delle funzioni renali parziali: trasporto di aminoacidi, elettroliti, funzione di concentrazione, acidogenesi. Le alterazioni successive influenzano lo stato funzionale sia delle porzioni prossimali che distali del nefrone e sono caratterizzate da disordini parziali combinati. Una riduzione della filtrazione glomerulare si verifica più tardi, più spesso nell'adolescenza. Con il progredire della nefrite ereditaria, si sviluppa anemia.
Pertanto, la nefrite ereditaria è caratterizzata da un decorso a stadi della malattia: inizialmente, una fase latente o sintomi clinici nascosti, manifestati da minime alterazioni della sindrome urinaria, poi si verifica un graduale scompenso del processo con una riduzione della funzionalità renale e sintomi clinici manifesti (intossicazione, astenia, ritardo dello sviluppo, anemia). I sintomi clinici di solito compaiono indipendentemente dalla stratificazione della reazione infiammatoria.
La nefrite ereditaria può manifestarsi in diversi periodi dell'età, a seconda dell'azione del gene, che fino a un certo momento si trova in stato represso.
Classificazione
Esistono tre tipi di nefrite ereditaria
- Opzione I - si manifesta clinicamente come nefrite con ematuria, perdita dell'udito e danni oculari. Il decorso della nefrite è progressivo con lo sviluppo di insufficienza renale cronica. Il tipo di ereditarietà è dominante, legata al cromosoma X. Morfologicamente, si evidenzia una violazione della struttura della membrana basale, con il suo assottigliamento e la sua scissione.
- Opzione II - si manifesta clinicamente come nefrite con ematuria senza perdita dell'udito. Il decorso della nefrite è progressivo con lo sviluppo di insufficienza renale cronica. Il tipo di ereditarietà è dominante, legato al cromosoma X. Morfologicamente, si rileva un assottigliamento della membrana basale dei capillari glomerulari (in particolare della laminadensa).
- Opzione III - ematuria familiare benigna. Il decorso è favorevole, non si sviluppa insufficienza renale cronica. La trasmissione è autosomica dominante o autosomica recessiva. Con la trasmissione autosomica recessiva, si osserva un decorso più grave della malattia nelle donne.
Diagnosi della sindrome di Alport
Vengono proposti i seguenti criteri:
- la presenza di almeno due pazienti con nefropatia in ogni famiglia;
- ematuria come sintomo principale di nefropatia nel probando;
- la presenza di perdita dell'udito in almeno un membro della famiglia;
- sviluppo di insufficienza renale cronica in uno o più parenti.
Nella diagnosi di varie malattie ereditarie e congenite, un ruolo importante è svolto da un approccio completo all'esame e, soprattutto, dall'attenzione ai dati ottenuti durante la compilazione dell'albero genealogico del bambino. La diagnosi di sindrome di Alport è considerata valida nei casi in cui vengano rilevati nel paziente 3 dei 4 segni tipici: presenza di ematuria e insufficienza renale cronica in famiglia, presenza di ipoacusia neurosensoriale, patologia visiva nel paziente, rilevamento di segni di clivaggio della membrana basale glomerulare con alterazione del suo spessore e contorni irregolari durante l'esame microscopico elettronico della biopsia.
L'esame obiettivo del paziente deve includere metodi di ricerca clinica e genetica; uno studio mirato dell'anamnesi; un esame obiettivo generale del paziente tenendo conto di criteri diagnostici significativi. Nella fase di compensazione, la patologia può essere rilevata solo concentrandosi su sindromi quali la presenza di un carico ereditario, l'ipotensione, i molteplici stigmi di disembriogenesi e le alterazioni della sindrome urinaria. Nella fase di scompenso, possono comparire sintomi extrarenali, come grave intossicazione, astenia, ritardo dello sviluppo fisico, anemia, che si manifestano e si intensificano con una graduale riduzione della funzionalità renale. Nella maggior parte dei pazienti, con una riduzione della funzionalità renale, si osserva quanto segue: riduzione dell'acidogenesi e dell'aminogenesi; il 50% dei pazienti nota una significativa riduzione della funzione secretoria renale; un intervallo limitato di fluttuazioni nella densità ottica delle urine; un disturbo del ritmo di filtrazione e, di conseguenza, una riduzione della filtrazione glomerulare. Lo stadio di insufficienza renale cronica viene diagnosticato quando i pazienti presentano un livello elevato di urea nel siero sanguigno (superiore a 0,35 g/l) per 3-6 mesi o più e una diminuzione della filtrazione glomerulare al 25% della norma.
La diagnosi differenziale della nefrite ereditaria dovrebbe essere eseguita principalmente con la forma ematurica della glomerulonefrite acquisita. La glomerulonefrite acquisita ha più spesso un esordio acuto, entro 2-3 settimane dall'infezione, segni extrarenali, tra cui ipertensione fin dai primi giorni (nella nefrite ereditaria, al contrario, ipotensione), ridotta filtrazione glomerulare all'esordio della malattia, nessuna compromissione delle funzioni tubulari parziali, mentre nella glomerulonefrite ereditaria sono presenti. La glomerulonefrite acquisita si manifesta con ematuria e proteinuria più pronunciate, con un aumento della VES. Le tipiche alterazioni della membrana basale glomerulare, caratteristiche della nefrite ereditaria, hanno valore diagnostico.
La diagnosi differenziale con la nefropatia dismetabolica viene effettuata in caso di insufficienza renale cronica, in presenza di patologie renali eterogenee clinicamente riscontrate in famiglia, e può essere presente uno spettro di nefropatie che va dalla pielonefrite all'urolitiasi. I bambini lamentano spesso dolore addominale e, periodicamente, durante la minzione, presenza di ossalati nel sedimento urinario.
Se si sospetta una nefrite ereditaria, il paziente deve essere indirizzato a un reparto di nefrologia specializzato per chiarire la diagnosi.
Cosa c'è da esaminare?
Come esaminare?
Quali test sono necessari?
Chi contattare?
Trattamento della sindrome di Alport
Il regime terapeutico prevede restrizioni sull'esercizio fisico intenso e sull'esposizione all'aria aperta. La dieta è completa, con livelli sufficienti di proteine, grassi e carboidrati completi, tenendo conto della funzionalità renale. Di grande importanza è l'individuazione e il trattamento dei focolai cronici di infezione. Vengono utilizzati i seguenti farmaci: ATP, cocarbossilasi, piridossina (fino a 50 mg/die), cloruro di carnitina. I cicli vengono somministrati 2-3 volte all'anno. Per l'ematuria, vengono prescritti rimedi erboristici: ortica, succo di aronia, achillea.
Esistono segnalazioni in letteratura nazionale e straniera sul trattamento con prednisolone e sull'uso di citostatici. Tuttavia, è difficile valutarne l'efficacia.
In caso di insufficienza renale cronica si ricorre all'emodialisi e al trapianto di rene.
Non esistono metodi di terapia specifici (patogenetici efficaci) per la nefrite ereditaria. Tutte le misure terapeutiche mirano a prevenire e rallentare il declino della funzionalità renale.
La dieta deve essere equilibrata e ipercalorica, tenendo conto dello stato funzionale dei reni. In assenza di disturbi funzionali, la dieta del bambino deve contenere sufficienti proteine, grassi e carboidrati. In presenza di segni di disfunzione renale, la quantità di proteine, carboidrati, calcio e fosforo deve essere limitata, il che ritarda lo sviluppo di insufficienza renale cronica.
L'attività fisica dovrebbe essere limitata; si consiglia ai bambini di evitare lo sport.
Si raccomanda di evitare il contatto con pazienti infettivi, riducendo il rischio di sviluppare malattie respiratorie acute. È necessaria la sanificazione dei focolai di infezione cronica. Le vaccinazioni preventive non vengono eseguite nei bambini con nefrite ereditaria; la vaccinazione è possibile solo per indicazioni epidemiologiche.
La terapia ormonale e immunosoppressiva nella nefrite ereditaria è inefficace. Esistono indicazioni di un effetto positivo (riduzione della proteinuria e rallentamento della progressione della malattia) con l'uso a lungo termine di ciclosporina A e ACE-inibitori per diversi anni.
Nel trattamento dei pazienti vengono utilizzati farmaci che migliorano il metabolismo:
- piridossina - 2-3 mg/kg/giorno in 3 dosi per 4 settimane;
- cocarbossilasi - 50 mg per via intramuscolare a giorni alterni, per un totale di 10-15 iniezioni;
- ATP - 1 ml per via intramuscolare a giorni alterni, 10-15 iniezioni;
- vitamina A - 1000 UI/anno/giorno in 1 dose per 2 settimane;
- Vitamina E - 1 mg/kg/giorno in 1 dose per 2 settimane.
Questo tipo di terapia aiuta a migliorare le condizioni generali dei pazienti, a ridurre le disfunzioni tubulari e viene eseguito a cicli 3 volte l'anno.
Il levamisolo può essere utilizzato come immunomodulatore: 2 mg/kg/giorno 2-3 volte alla settimana con pause tra le dosi di 3-4 giorni.
Secondo i dati della ricerca, l'ossigenazione iperbarica ha un effetto positivo sulla gravità dell'ematuria e della disfunzione renale.
Il metodo più efficace per trattare la nefrite ereditaria è il trapianto renale tempestivo. In questo caso, non si verifica alcuna recidiva della malattia durante il trapianto; in una piccola percentuale di casi (circa il 5%), la nefrite può svilupparsi nel rene trapiantato associata ad antigeni della membrana basale glomerulare.
Una direzione promettente è la diagnosi prenatale e la terapia di ingegneria genetica. Gli esperimenti sugli animali mostrano un'elevata efficienza nel trasferimento dei geni normali responsabili della sintesi delle catene alfa del collagene di tipo IV nel tessuto renale, dopodiché si osserva la sintesi di strutture di collagene normali.
Previsione
La prognosi della nefrite ereditaria è sempre grave.
I criteri prognosticamente sfavorevoli per il decorso della nefrite ereditaria sono:
- genere maschile;
- sviluppo precoce di insufficienza renale cronica nei membri della famiglia;
- proteinuria (più di 1 g/giorno);
- ispessimento delle membrane basali glomerulari secondo microscopia;
- neurite acustica;
- delezione nel gene Col4A5.
La prognosi dell'ematuria familiare benigna è più favorevole.
Использованная литература