Esperto medico dell'articolo

Nuove pubblicazioni
Displasia renale
Ultima recensione: 04.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
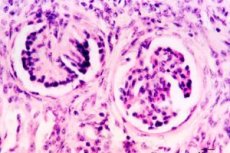
La displasia renale occupa un posto di rilievo tra i difetti dello sviluppo dell'apparato urinario. La displasia renale è un gruppo eterogeneo di malattie associate a un alterato sviluppo del tessuto renale. Morfologicamente, la displasia si basa su un'alterata differenziazione del blastema nefrogenico e dei rami dello stelo ureterale, con la presenza di strutture embrionali sotto forma di focolai di mesenchima indifferenziato, nonché di dotti e tubuli primitivi. Il mesenchima, rappresentato da cellule cambiali pluripotenti e fibre collagene, può formare derivati disontogenetici di cartilagine ialina e fibre muscolari lisce.
 [ 1 ]
[ 1 ]
Le cause displasia renale
Patogenesi
L'esame morfologico della displasia ipoplasica rivela una certa riduzione della massa renale, una superficie lobulare, una divisione in strati non sempre nettamente definita, a volte una certa espansione o ipoplasia degli ureteri. Microscopicamente, si rilevano strutture primitive: molti glomeruli sono ridotti di dimensioni, le anse vascolari sono atrofiche, la capsula è ispessita. La forma dei glomeruli può essere a S o ad anello, molti di essi sono ialinizzati e sclerosati. I glomeruli sono disposti a grappolo, circondati da tessuto connettivo lasso con accumuli focali di cellule linfoidi e istiocitarie. Nella midollare sono presenti numerosi dotti e tubuli primitivi, che sono formazioni immature di vari stadi dello sviluppo embrionale. I dotti primitivi si rilevano principalmente nella zona iuxtamidollare e sono i resti del dotto mesonefrogenico. Una caratteristica è la presenza di ombre di cellule muscolari lisce e fibre di tessuto connettivo attorno ad essi. La presenza di strutture primitive riflette un ritardo nella maturazione del nefrone.
L'esame morfologico della displasia focale semplice non rivela alterazioni significative della massa renale. In alcuni casi, si osserva una riduzione dello spessore della corticale. Questa nefropatia viene diagnosticata sulla base delle alterazioni istologiche rilevate al microscopio. La displasia focale semplice è caratterizzata dalla presenza di gruppi di glomeruli e tubuli primitivi, circondati da fibre di tessuto connettivo e cellule muscolari lisce, principalmente nella corticale renale; talvolta si riscontra tessuto cartilagineo. È caratteristico il polimorfismo dell'epitelio dei tubuli contorti, dove le cellule adiacenti differiscono per dimensioni, configurazione, disposizione e numero di organelli intracellulari. Alcuni bambini possono presentare lumi tubulari dilatati nei reni. Possono essere rilevate anche cisti glomerulari, ma il loro numero è insignificante. Le cellule mononucleate mesenchimali vengono determinate nello stroma.
La displasia segmentale semplice (rene di Ask-Upmark) è piuttosto rara (0,02% di tutte le autopsie). In questo tipo di displasia, il rene è di dimensioni ridotte, un solco trasversale è chiaramente visibile sulla superficie esterna in corrispondenza del segmento ipoplasico e il numero di piramidi è ridotto. Le alterazioni morfologiche sono causate dalla disembriogenesi dei vasi nei singoli segmenti renali, con conseguente interruzione della differenziazione delle strutture tissutali dovuta a variazioni dell'afflusso sanguigno a queste aree. Di solito si riscontra un iposviluppo dei corrispondenti rami arteriosi. Un segno caratteristico è la presenza di dotti mesonefogenici primitivi nel segmento ipoplasico, circondati da cellule muscolari lisce e focolai di cartilagine ialina. Inoltre, si sviluppano sclerosi, ialinosi glomerulare, atrofia dell'epitelio tubulare con dilatazione del lume, segni di fibrosi e infiltrazione cellulare e interstizio.
La displasia cistica aplastica (rene rudimentale multicistico) rappresenta il 3,5% di tutte le malformazioni congenite dell'apparato urinario e il 19% di tutte le forme di displasia cistica. I reni sono significativamente ridotti di dimensioni, sono formazioni cistiche informi di 2-5 mm di diametro, il parenchima renale è quasi completamente assente, l'uretere è assente o è presente atresia. Microscopicamente, si rileva un gran numero di cisti, sia glomerulari che tubulari, nonché dotti primitivi e focolai di tessuto cartilagineo. Il danno bilaterale è incompatibile con la vita. Il rene rudimentale monolaterale viene spesso rilevato durante un esame casuale e il secondo rene è spesso anomalo.
La displasia cistica ipoplasica (rene ipoplasico multicistico) rappresenta il 3,9% di tutti i difetti dell'apparato urinario e il 21,2% delle displasie cistiche. I reni sono ridotti in dimensioni e peso. Le cisti glomerulari si trovano solitamente nella zona sottocapsulare, il loro diametro varia e può raggiungere i 3-5 mm. Le cisti tubulari si trovano sia nella corticale che nella midollare. La fibrosi del tessuto connettivo e la presenza di dotti primitivi sono più significative nella midollare. Le cisti sono di grandi dimensioni e rappresentano tubuli collettori cisticamente dilatati. Il parenchima renale è parzialmente conservato. Tra le aree patologicamente alterate sono presenti tubuli collettori di normale struttura. La pelvi renale può essere invariata, più spesso ipoplasica, così come l'uretere. La displasia cistica ipoplasica è spesso associata a difetti del tratto urinario inferiore, del tratto gastrointestinale, del sistema cardiovascolare e di altri organi.
Il danno bilaterale porta precocemente allo sviluppo di insufficienza renale cronica. Di norma, nella variante monolaterale di questa displasia, il secondo rene presenta alcune manifestazioni di disembriogenesi.
La displasia cistica iperplastica accompagna spesso la sindrome di Patau. Il processo è bilaterale. I reni sono aumentati di volume e ricoperti da cisti multiple. L'esame microscopico rivela dotti primitivi e cisti in gran numero nella corticale e nella midollare. L'esito letale si verifica solitamente in età precoce.
La displasia multicistica (rene multicistico) è un difetto dello sviluppo in cui i reni risultano aumentati di dimensioni, è presente un gran numero di cisti di varie forme e dimensioni (da 5 mm a 5 cm), tra le quali il parenchima è praticamente assente.
L'esame microscopico rivela la presenza di dotti e glomeruli primitivi tra le cisti, e possono essere presenti anche aree con tessuto cartilagineo. In caso di lesioni bilaterali, la morte avviene nei primi giorni di vita. In caso di lesioni monolaterali, la diagnosi viene posta casualmente durante la palpazione di una formazione simil-tumorale tuberosa o in base ai risultati ecografici. In caso di malattia multicistica monolaterale, possono essere presenti malformazioni a carico del secondo rene (spesso idronefrosi), difetti cardiaci, difetti del tratto gastrointestinale, ecc.
Nella displasia midollare (displasia cistica della midollare, malattia cistica midollare, nefronoftisi di Fanconi), i reni sono solitamente di dimensioni ridotte, spesso mantenendo la lobulazione embrionale. La corticale è assottigliata, la midollare è espansa a causa di un gran numero di cisti fino a 1 cm di diametro, inclusa la caratteristica espansione cistica dei dotti collettori. L'esame microscopico rivela una riduzione delle dimensioni di molti glomeruli, alcuni dei quali sono ialinizzati e sclerotici; anche l'interstizio è sclerotico e vi è infiltrazione linfoide nello stroma.
Un posto speciale tra le displasie cistiche è occupato dalla malattia renale policistica. La comparsa della malattia renale policistica è associata a un disturbo dello sviluppo embrionale dei reni, il più delle volte dovuto a una mancanza di connessione dei tubuli collettori primari con una parte del nefrone che si sviluppa da un blastoma metanefrogenico. I tubuli ciechi formatisi in questo caso continuano a svilupparsi, l'urina primaria si accumula al loro interno, distendendoli e causando atrofia epiteliale. Allo stesso tempo, il tessuto connettivo che circonda i tubuli cresce.
Le dimensioni delle cisti variano ampiamente: oltre a quelle piccole, visibili solo con una lente d'ingrandimento o persino al microscopio, ce ne sono di grandi dimensioni, fino a diversi centimetri di diametro. Un gran numero di cisti a parete sottile nella corticale e nella midollare dei reni conferisce loro l'aspetto di un nido d'ape al taglio. Istologicamente, le cisti sono rappresentate da tubuli dilatati con epitelio cubico o hanno l'aspetto di cavità con una parete di tessuto connettivo spesso ed epitelio nettamente appiattito. E. Potter (1971) ha descritto cisti associate all'espansione della cavità della capsula di Bowman dei glomeruli, senza alterazioni dei tubuli. Le cisti possono essere vuote o contenere liquido sieroso e proteico, a volte colorato con pigmenti ematici e cristalli di acido urico. Lo stroma dei reni nella malattia policistica è sclerotico, spesso con infiltrazione focale di cellule linfoidi e, nei bambini di età inferiore a 1 anno, con focolai di emopoiesi extramidollare. Talvolta si trovano isole di cartilagine o fibre muscolari lisce nello stroma. Il numero e il tipo di glomeruli e tubuli situati tra le cisti possono variare.
Sintomi displasia renale
La displasia totale semplice è spesso descritta in letteratura come displasia ipoplasica. Tra tutte le malformazioni congenite dell'apparato urinario, rappresenta il 2,7%.
Si distingue tra varianti aplastiche e ipoplastiche. Nel caso della displasia renale aplastica, in caso di lesioni bilaterali, la morte avviene nelle prime ore o giorni di vita.
La variante ipoplastica è caratterizzata dalla manifestazione precoce della sindrome urinaria, caratterizzata da mosaicismo, e dallo sviluppo precoce di insufficienza renale cronica.
La displasia focale semplice viene solitamente diagnosticata mediante nefrobiopsia o autopsia. Non vi sono manifestazioni cliniche della malattia.
Nella displasia segmentale semplice, il sintomo dominante è lo sviluppo di ipertensione arteriosa persistente già in età precoce, più comune nelle bambine. I bambini lamentano mal di testa, possono manifestarsi convulsioni e si sviluppano precocemente alterazioni dei vasi del fondo oculare.
Uno dei principali sintomi clinici è la sindrome dolorosa, che si manifesta con dolore addominale, poliuria e polidipsia, che si manifestano abbastanza precocemente come manifestazioni della sindrome tubulo-interstiziale. In alcuni casi, si osserva un ritardo nel peso corporeo e nella crescita dei bambini. La sindrome urinaria si manifesta con proteinuria predominante, associata a microematuria e leucocituria moderata.
I segni clinici della malattia renale policistica compaiono nell'adolescenza: dolore lombare, palpazione di una formazione simil-tumorale nella cavità addominale, ipertensione arteriosa. La sindrome urinaria si manifesta con ematuria. Spesso si associa pielonefrite. Dal punto di vista funzionale, i reni sono conservati per molti anni, poi compaiono ipostenuria, ridotta filtrazione glomerulare e iperazotemia.
La cisti multiloculare (displasia cistica focale del rene) è una forma focale di displasia cistica del rene ed è caratterizzata dalla presenza di una cisti multicamerale in uno dei suoi poli, delimitata da una capsula di tessuto renale normale e divisa internamente da setti.
Il quadro clinico di una cisti multiloculare è caratterizzato dalla comparsa di una sindrome dolorosa di varia intensità a livello addominale e lombare, dovuta all'interruzione del deflusso urinario dovuta alla compressione della pelvi renale o dell'uretere da parte di una cisti di grandi dimensioni. Inoltre, a causa della possibile compressione degli organi addominali, si manifestano sintomi che simulano la patologia.
Le manifestazioni cliniche della displasia midollare si sviluppano solitamente dopo i 3 anni di età, più spesso all'età di 5-6 anni compare il "complesso di sintomi di Fanconi": poliuria, polidipsia, aumento della temperatura corporea, ritardo dello sviluppo fisico, vomito ripetuto, disidratazione, acidosi, anemia, rapida progressione dell'uremia.
Il quadro clinico della displasia cistica aplastica è determinato dalle condizioni del secondo rene, nel quale spesso si sviluppa pielonefrite a causa della presenza di displasia.
La displasia multicistica può manifestarsi con la presenza di dolore sordo o parossistico all'addome e alla regione lombare. Può essere riscontrata ipertensione arteriosa.
Nella displasia corticale (malattia renale microcistica, sindrome nefrosica congenita di tipo "finlandese"), le dimensioni dei reni non sono alterate e la lobulazione può essere conservata. Si riscontrano piccole cisti glomerulari e tubulari con un diametro di 2-3 mm. Il quadro clinico della sindrome nefrosica si osserva fin dalla nascita. La sindrome nefrosica congenita di tipo "finlandese" è ormono-resistente e ha una prognosi sfavorevole. Si osserva uno sviluppo precoce di insufficienza renale cronica.
Il quadro clinico della displasia cistica ipoplasica è causato dalla pielonefrite, lo sviluppo di un'insufficienza renale cronica, la cui velocità di progressione dipende non solo dalla quantità di parenchima conservato del rene ipoplasico, ma anche dal grado di danno al secondo rene non ipoplasico, ma, di regola, presentante elementi displastici.
La displasia ipoplastica può essere rilevata sullo sfondo di una malattia intercorrente, mentre le sindromi extrarenali possono essere assenti o debolmente espresse. La sindrome urinaria si manifesta con ematuria con proteinuria moderata. Le manifestazioni di questa malattia sono molto eterogenee. Spesso può essere presente una variante proteinurica con significativa perdita di proteine, ma la sindrome edematosa è relativamente rara, anche con proteinuria significativa, e la sindrome nefrosica è caratterizzata come incompleta. L'osservazione dinamica del bambino mostra che il quadro clinico è successivamente caratterizzato da sindrome nefrosica, dalla presenza di alterazioni tubulo-interstiziali, spesso con la stratificazione di un'infezione del tratto urinario.
I bambini con displasia ipoplastica sviluppano tipicamente stati ipoimmuni o di immunodeficienza, il che spiega l'aggiunta di gravi e frequenti malattie intercorrenti con la progressione del processo patologico renale. Una caratteristica importante di questa nefropatia è l'assenza di ipertensione arteriosa; l'ipotensione è più comune. Un aumento della pressione arteriosa si verifica già con lo sviluppo di insufficienza renale cronica.
Il decorso della displasia ipoplastica è torpore, non vi è ciclicità o natura ondulatoria delle manifestazioni, la terapia farmacologica è solitamente inefficace.
Forme
Attualmente, non esiste una classificazione generalmente accettata della displasia renale. La maggior parte degli autori, in base alle manifestazioni morfologiche, distingue tra displasie semplici e cistiche e, in base alla localizzazione, corticale, midollare e corticomidollare. A seconda della prevalenza, si distinguono displasie focali, segmentali e totali.
A seconda della prevalenza, si distinguono forme totali, focali e segmentali di displasia cistica.
Tra le forme totali di displasia cistica si distinguono le varianti aplastica, ipoplastica, iperplastica e multicistica.
La malattia policistica si manifesta in due forme principali, che differiscono per la natura dell'ereditarietà, le manifestazioni cliniche e il quadro morfologico: la forma "infantile" e quella "adulta".
La malattia policistica di tipo "infantile" (rene cistico piccolo) ha una trasmissione autosomica recessiva. I reni sono significativamente aumentati di dimensioni e peso. Numerose cisti cilindriche e fusiformi sono presenti nella corticale e nella midollare. Le cisti sono delimitate da scarsi strati di tessuto connettivo. Le cisti si trovano anche nel fegato e in altri organi. Le manifestazioni cliniche dipendono dal numero di tubuli interessati. Con un danno al 60% dei tubuli, la morte per uremia progressiva si verifica nei primi 6 mesi. I risultati di O. V. Chumakova (1999) non confermano i concetti classici di mortalità precoce nei bambini con malattia policistica autosomica recessiva e dimostrano che la loro aspettativa di vita può essere piuttosto lunga, anche con una diagnosi precoce dei sintomi clinici. Tuttavia, l'insufficienza renale cronica si sviluppa prima in questi pazienti rispetto alla forma autosomica dominante della malattia policistica. In questi pazienti, il ruolo principale nel quadro clinico è svolto dai sintomi di danno epatico. Micro-, macroematuria e aumento della pressione arteriosa sono spesso osservati in clinica. La malattia policistica è spesso complicata da pielonefrite con decorso torpido.
Nella malattia policistica dell'adulto (rene cistico di grandi dimensioni), i reni sono quasi sempre di dimensioni aumentate, raggiungendo negli adulti un peso di 1,5 kg o più ciascuno. Nella corticale e nella midollare sono presenti numerose cisti fino a 4-5 cm di diametro.
Diagnostica displasia renale
La diagnosi della malattia renale policistica si basa sull'anamnesi familiare, sui dati ecografici, sull'urografia escretoria, che mostrano un aumento dei contorni dei reni, un appiattimento della pelvi renale con allungamento, dilatazione e compressione dei calici.
Nella diagnosi delle cisti multiloculari, i metodi di esame radiologico, tra cui la nefrotomografia e l'angiografia, sono di importanza decisiva.
Tra i segni di laboratorio della displasia midollare, l'ipoproteinemia è caratteristica; la sindrome urinaria si manifesta solitamente con una lieve proteinuria. A causa dell'aumentata perdita di sali, si sviluppano iponatriemia, ipokaliemia e ipocalcemia. L'acidosi si sviluppa a causa di una significativa bicarbonaturia, una compromissione dell'acidogenesi e dell'ammoniogenesi.
La diagnosi di displasia cistica aplastica si basa su ecografie, urografia escretoria, nefroscopia e scintigrafia. Durante la cistoscopia, l'orifizio ureterale sul lato del rene rudimentale è solitamente assente o stenotico.
Per la diagnosi di displasia ipoplastica, sono di grande importanza la scoperta accidentale della malattia, la presenza di molteplici stigmi di disembriogenesi e un certo ritardo nello sviluppo fisico.
Come esaminare?
Quali test sono necessari?
Diagnosi differenziale
Trattamento displasia renale
Il trattamento della displasia ipoplastica è sintomatico.
Se viene diagnosticata la malattia multicistica, si procede con la nefrectomia, a causa del rischio di sviluppare tumori maligni.
Il trattamento della displasia midollare è sintomatico. In caso di insufficienza renale cronica, sono indicati l'emodialisi o la dialisi peritoneale e il trapianto renale.
Previsione
La prognosi della displasia ipoplastica è grave, con sviluppo precoce di insufficienza renale cronica e necessità di terapia sostitutiva: emodialisi o dialisi peritoneale, trapianto di rene.
[ 28 ]
Использованная литература