Esperto medico dell'articolo

Nuove pubblicazioni
Neuroblastoma nei bambini: cause, diagnosi, trattamento
Ultima recensione: 04.07.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
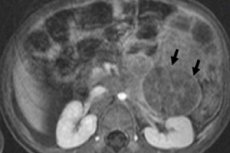
In oncologia pediatrica, una delle neoplasie extracraniche più comuni nei bambini è il neuroblastoma, un tumore maligno embrionale dei neuroblasti della cresta neurale, cioè le cellule nervose embrionali (immature) del sistema nervoso simpatico.
Epidemiologia
Secondo le statistiche dell'International Neuroblastoma Risk Group (INRG), il neuroblastoma rappresenta circa l'8% di tutte le malattie oncologiche infantili in tutto il mondo ed è il terzo per prevalenza, dopo la leucemia e i tumori cerebrali.
Secondo altri dati, il neuroblastoma rappresenta circa il 28% di tutti i tumori infantili. Oltre un terzo dei casi di neuroblastoma viene diagnosticato in bambini di età inferiore a un anno; l'età media della diagnosi è di 19-22 mesi. Oltre il 90% dei casi diagnosticati si verifica in bambini di età compresa tra due e cinque anni (con una predominanza di maschi); il picco di incidenza si osserva tra i due e i tre anni, e i casi nei bambini di età superiore ai cinque anni rappresentano meno del 10%.
Le cause neuroblastomi
Studiando le cause del neuroblastoma, i ricercatori hanno concluso che questo tumore nei bambini si verifica a causa di mutazioni genetiche sporadiche durante l'embriogenesi o lo sviluppo postnatale precoce. Tuttavia, le cause di queste alterazioni genetiche sono sconosciute, poiché non è stata identificata alcuna influenza di fattori ambientali teratogeni.
Questi tumori possono presentarsi ovunque, tra cui mediastino, collo, addome, ghiandole surrenali, reni, colonna vertebrale e bacino.
In rari casi, il neuroblastoma infantile può essere associato a una mutazione ereditaria. In particolare, una mutazione nel gene della proteina di membrana CD246 sul cromosoma 2 - l'enzima tirosin-chinasi ALK, che assicura le comunicazioni intercellulari e svolge un ruolo importante nel funzionamento del sistema nervoso - e nel gene della proteina PHOX2B (sul cromosoma 4), coinvolta nella maturazione delle cellule nervose.
Il neuroblastoma può anche essere associato alla neurofibromatosi infantile di tipo 1,alla sindrome di Beckwith-Wiedemann e all'ipoglicemia iperinsulinemica (pancreatite nesidioblastosi).
Fattori di rischio
Oggi, l'ereditarietà è riconosciuta come fattore di rischio per lo sviluppo del neuroblastoma nei bambini: la presenza di questo tumore nella storia familiare, così come anomalie congenite associate a mutazioni genetiche durante lo sviluppo intrauterino. Ciò è particolarmente vero nei casi di sviluppo di diverse neoplasie in organi diversi.
Nessuno dei fattori esogeni che aumentano il rischio di questo tumore è stato identificato dai ricercatori.
Patogenesi
Il meccanismo di sviluppo dei neuroblastomi è causato da anomalie nella differenziazione e maturazione delle cellule della cresta neurale, linee cellulari bilaterali che si formano ai margini del tubo neurale a partire dal foglietto embrionale ectodermico dell'embrione umano. Queste cellule migrano (si muovono) e si differenziano in numerosi tipi di cellule: neuroni sensoriali e autonomici, cellule neuroendocrine e cellule della midollare surrenale, cellule della cartilagine e delle ossa craniofacciali, nonché cellule pigmentate.
Nel neuroblastoma, i neuroblasti migrati non maturano, ma continuano a crescere e dividersi, formando un tumore. La patogenesi della sua formazione è associata alle seguenti mutazioni genetiche:
- con duplicazione di parte della sequenza cromosomica o duplicazione di segmenti del gene LMO1 sul cromosoma 11, che codifica la proteina RBTN1 nelle cellule della cresta neurale dell'embrione;
- con un'alterazione del numero di copie del gene NBPF10 sul cromosoma 1q21.1, che codifica per la proteina DUF1220, che controlla la proliferazione delle cellule staminali neurali umane. Queste patologie portano alla duplicazione di questo cromosoma o alla sua delezione, ovvero all'assenza di una parte del DNA;
- con alterazioni del gene soppressore del tumore ATRX (sul cromosoma Xq21.1);
- con la presenza di copie aggiuntive (amplificazione) del gene del fattore di trascrizione N-Myc sul cromosoma 2, che codifica per uno dei fattori di trascrizione (proteina legante il DNA) che regola l'attività di altri geni e controlla la proliferazione delle cellule precursori durante la formazione di proteine per la formazione di tessuti e organi del feto. L'amplificazione di questo gene lo trasforma in un oncogene, provocando un'interruzione del ciclo cellulare, un aumento della proliferazione cellulare e la formazione di tumori.
Sintomi neuroblastomi
I primi segni del neuroblastoma sono aspecifici e possono includere perdita di appetito (e perdita di peso), affaticamento durante l'alimentazione, febbre e dolori articolari.
I sintomi clinici dipendono dalla sede del tumore primario e dalla presenza di metastasi (presenti nel 60-73% dei casi).
Molto spesso, il neuroblastoma primario è localizzato nella midollare del surrene, che ha un'origine simile alle cellule nervose. Nei bambini di età inferiore a un anno, il neuroblastoma surrenalico viene diagnosticato nel 35-40% dei casi. I suoi sintomi includono dolore addominale, febbre, perdita di peso, dolore osseo, anemia o concomitante sindrome di Pepper: danno epatico diffuso con grave epatomegalia e sindrome da distress respiratorio.
Il neuroblastoma retroperitoneale o neuroblastoma retroperitoneale nei bambini, crescendo, inizia a premere sulla vescica o sull'intestino, il che può causare problemi di minzione o defecazione, gonfiore delle gambe (nei ragazzi, si gonfia lo scroto).
Il neuroblastoma del mediastino nei bambini (neuroblastoma mediastinico) spesso comprime la vena cava superiore e questo può causare gonfiore di viso, collo, braccia e parte superiore del torace (con la pelle che diventa bluastra-rossastra, con noduli sottocutanei). Compaiono tosse e respiro sibilante, problemi respiratori (dispnea) o difficoltà di deglutizione (disfagia); si notano linfonodi ingrossati nel collo, sopra la clavicola e sotto le ascelle.
La diffusione delle cellule tumorali al midollo osseo provoca anemia, trombocitopenia e leucopenia con tendenza al sanguinamento.
In caso di metastasi nella zona periorbitale, compaiono occhiaie o lividi intorno agli occhi. Un tumore di questo tipo può anche causare mal di testa e vertigini, esoftalmo (gonfiore dei bulbi oculari) e, a causa della compressione delle terminazioni nervose, palpebre cadenti (ptosi) e riduzione delle dimensioni delle pupille (miosi).
Il neuroblastoma addominale o neuroblastoma della cavità addominale nei bambini porta alla formazione di suggelli palpabili nell'addome, alla sua distensione, alla perdita di appetito, alla stitichezza e all'aumento della pressione sanguigna. Un tumore che preme sul midollo spinale o sulle radici nervose può causare intorpidimento e debolezza degli arti, incapacità di stare in piedi, gattonare o camminare. Se le ossa sono colpite, può verificarsi dolore osseo.
In caso di tumore allo stadio 3-4 nella cavità addominale con danni ai linfonodi, le cellule tumorali possono penetrare nel parenchima renale e quindi si sviluppa un esteso neuroblastoma del rene nei bambini, con conseguente compromissione delle sue funzioni.
Fasi
- Il neuroblastoma allo stadio 1 è un tumore primario localizzato e isolato in una zona del corpo; i linfonodi su entrambi i lati non sono interessati.
- Neuroblastoma stadio 2. Nello stadio 2A, il tumore primario è confinato a un'area ma è di grandi dimensioni; i linfonodi bilaterali non sono interessati. Nello stadio 2B, i linfonodi del lato del corpo in cui è localizzato il tumore sono positivi per metastasi.
- Neuroblastoma stadio 3: il tumore primario attraversa il midollo spinale o la linea mediana del corpo, nei linfonodi si riscontrano metastasi unilaterali o bilaterali.
- Neuroblastoma stadio 4: il tumore si è diffuso a linfonodi distanti, midollo osseo, ossa, fegato o altri organi. Lo stadio 4S si riscontra nei bambini di età inferiore a un anno con tumore primario localizzato, con disseminazione alla cute, al fegato o al midollo osseo.
Sistema internazionale di stadiazione del rischio di neuroblastoma (INRGSS)
L'INRGSS utilizza i fattori di rischio definiti dall'imaging (IDRF), ovvero fattori rilevati negli esami di diagnostica per immagini che potrebbero rendere più difficile la rimozione di un tumore.
L'INRGSS divide i neuroblastomi in 4 stadi:
- L1: Il tumore non si è diffuso dalla sede di origine e non ha raggiunto strutture vitali. È limitato a una parte del corpo, come collo, torace o addome.
- L2: Il tumore non si è diffuso (metastatizzato) lontano dal punto di origine (ad esempio, potrebbe essere cresciuto dal lato sinistro dell'addome al lato sinistro del torace), ma presenta almeno un IDRF.
- M: Il tumore ha metastatizzato in una parte distante del corpo (ad eccezione dei tumori allo stadio SM).
- SM: Malattia metastatica nei bambini di età inferiore ai 18 mesi, in cui il cancro si è diffuso solo alla pelle, al fegato e/o al midollo osseo.
Complicazioni e conseguenze
Il neuroblastoma è caratterizzato da complicazioni e conseguenze quali:
- diffusione (metastasi) ai linfonodi, al midollo osseo, al fegato, alla pelle e alle ossa;
- compressione del midollo spinale (che può causare dolore e portare alla paralisi);
- sviluppo della sindrome paraneoplastica (dovuta all'azione di alcune sostanze chimiche secrete dal tumore, nonché dell'antigene disialoganglioside GD2 espresso dalle sue cellule), che si manifesta con rapidi movimenti oculari involontari, disturbi della coordinazione, crampi muscolari e diarrea;
- ricadute dopo il completamento della terapia primaria (come dimostra la pratica clinica, i neuroblastomi ad alto rischio presentano una recidiva nel 50% dei casi).
Diagnostica neuroblastomi
Per diagnosticare un sospetto neuroblastoma in un bambino sono necessari esami, esami di laboratorio e diagnostica per immagini.
Vengono eseguiti esami del sangue e delle urine per la ricerca di catecolamine (noradrenalina e dopamina) e di acidi omovanillico o vanilmandelico (formati durante il metabolismo di questi ormoni); un esame del sangue per l'enolasi neurospecifica, un test immunoenzimatico (ELISA) del siero e un'analisi del midollo osseo (un campione del quale viene prelevato tramite agoaspirato). Viene eseguito un test del DNA per determinare le mutazioni e una biopsia per l'esame citomorfologico del tessuto tumorale.
Dopo il prelievo dei campioni bioptici, questi vengono inviati in laboratorio dove vengono esaminati al microscopio da un patologo (un medico specializzato nell'identificazione delle cellule tumorali). Spesso vengono eseguiti anche esami di laboratorio specifici sui campioni per determinare se il tumore è neuroblastoma.
Se si tratta di neuroblastoma, gli esami di laboratorio possono anche aiutare a stabilire la velocità con cui il tumore potrebbe crescere o diffondersi, nonché quali trattamenti potrebbero essere più efficaci.
La diagnostica strumentale visualizza la neoplasia mediante ecografia, radiografia, risonanza magnetica o TC, PET con l'introduzione della scintigrafia con 18F-fluorodesossiglucosio o MIBG con metaiodobenzilguanidina. [ 1 ]
Diagnosi differenziale
La diagnosi differenziale include il ganglioneuroma benigno, il ganglioneuroblastoma, il rabdomiosarcoma e il nefroblastoma.
Chi contattare?
Trattamento neuroblastomi
Nel neuroblastoma, il trattamento dipende dal gruppo di rischio del paziente (stadio del processo tumorale), dalla localizzazione del tumore, dalle caratteristiche genomiche delle cellule tumorali e dall'età del bambino. Può includere monitoraggio, intervento chirurgico, chemioterapia, radioterapia, immunoterapia e trapianto di cellule staminali emopoietiche.
La chemioterapia neoadiuvante o adiuvante (pre o postoperatoria) per il neuroblastoma pediatrico, come qualsiasi chemioterapia contro il cancro, viene somministrata in cicli: il farmaco viene somministrato per diversi giorni consecutivi, seguiti da una pausa per consentire all'organismo di recuperare. I cicli vengono solitamente ripetuti ogni tre o quattro settimane.
Vengono utilizzati i seguenti farmaci (e le loro combinazioni): Ciclofosfamide, Cisplatino o Carboplatino, Doxorubicina (Adriamicina), Vincristina, Etoposide.
Gli effetti collaterali comuni dei farmaci chemioterapici includono perdita di capelli, perdita di appetito, affaticamento, nausea e vomito, ulcere della bocca, diarrea o stitichezza. La chemioterapia può danneggiare il midollo osseo e causare una diminuzione della conta delle cellule del sangue.
L'immunoterapia mirata (diretta contro l'antigene tumorale GD2) utilizza farmaci appartenenti al gruppo degli anticorpi monoclonali (MAb anti-GD2) Dinutuximab (Unituxin) e Naxitamab. Vengono somministrati per via endovenosa per infusione prolungata, in combinazione con il fattore stimolante le colonie di granulociti-macrofagi (citochina GM-CSF) e l'interleuchina-2.
Gli effetti collaterali di questi farmaci comprendono dolore (spesso molto forte), diminuzione della pressione sanguigna, aumento della frequenza cardiaca, mancanza di respiro (con possibile gonfiore delle vie aeree), aumento della temperatura, nausea, vomito e diarrea, alterazioni nella composizione cellulare e minerale del sangue.
Per ridurre il rischio di recidiva del cancro dopo chemioterapia ad alte dosi e trapianto di cellule staminali, i bambini con neuroblastoma ad alto rischio vengono trattati con retinoidi sistemici, acido 13-cis-retinoico (isotretinoina). [ 2 ]
Trattamento chirurgico del neuroblastoma – rimozione del tumore, ad esempio, surrenectomia aperta o resezione laparoscopica del neuroblastoma surrenale; linfectomia (rimozione dei linfonodi interessati), ecc. [ 3 ]
Per il neuroblastoma ad alto rischio, può essere utilizzata la radioterapia.[ 4 ]
Prevenzione
Date le cause del neuroblastoma nei bambini, l'unica misura preventiva potrebbe essere la consulenza genetica al momento della pianificazione di una gravidanza. Tuttavia, è importante tenere presente che questo tumore è associato a mutazioni ereditarie solo nell'1-2% dei casi.
Previsione
Il neuroblastoma infantile ha la capacità di regredire spontaneamente.
Marcatori prognostici
- I tumori ad alto rischio, così come il neuroblastoma nei bambini di tutte le fasce d'età e di tutti gli stadi (tranne lo stadio 4S) – con aumentata espressione del gene N-MYC e amplificazione dell'oncogene N-Myc – hanno una prognosi sfavorevole che influisce sull'aspettativa di vita.
- Avere cellule tumorali che mancano di alcune parti dei cromosomi 1 o 11 (note come delezioni 1p o 11q) ha una prognosi peggiore. Anche avere una parte in più del cromosoma 17 (guadagno del cromosoma 17q) è associato a una prognosi peggiore.
- Le cellule del neuroblastoma con una grande quantità di DNA hanno una prognosi migliore, soprattutto nei bambini di età inferiore ai 2 anni.
- I neuroblastomi che presentano più recettori per le neurotrofine, in particolare il recettore del fattore di crescita nervoso TrkA, hanno una prognosi migliore.
Sopravvivenza per gruppo di rischio del Childhood Oncology Group (COG)
- Gruppo a basso rischio: i bambini appartenenti al gruppo a basso rischio hanno un tasso di sopravvivenza a 5 anni superiore al 95%.
- Gruppo a rischio intermedio: i bambini appartenenti al gruppo a rischio intermedio hanno un tasso di sopravvivenza a 5 anni dal 90% al 95%.
- Gruppo ad alto rischio: i bambini appartenenti al gruppo ad alto rischio hanno un tasso di sopravvivenza a 5 anni di circa il 50%.
Circa il 15% dei decessi per cancro infantile è dovuto al neuroblastoma. Le probabilità di sopravvivenza a lungo termine per questa neoplasia ad alto rischio non superano il 40%. Il tasso di sopravvivenza complessivo a cinque anni è del 67-74%, del 43% nella fascia di età da uno a quattro anni e di oltre l'80% per il neuroblastoma diagnosticato nel primo anno di vita.
Использованная литература