Esperto medico dell'articolo

Nuove pubblicazioni
amartoma
Ultima recensione: 29.06.2025

Tutti i contenuti di iLive sono revisionati o verificati da un punto di vista medico per garantire la massima precisione possibile.
Abbiamo linee guida rigorose in materia di sourcing e colleghiamo solo a siti di media affidabili, istituti di ricerca accademici e, ove possibile, studi rivisti dal punto di vista medico. Nota che i numeri tra parentesi ([1], [2], ecc.) Sono link cliccabili per questi studi.
Se ritieni che uno qualsiasi dei nostri contenuti sia impreciso, scaduto o comunque discutibile, selezionalo e premi Ctrl + Invio.
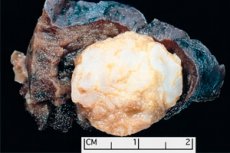
Formazione tumorale localizzata in qualsiasi regione anatomica derivante da una crescita anomala di tessuto benigno, in medicina è definita amartoma (dal greco hamartia - errore, difetto). [ 1 ]
Epidemiologia
Statisticamente, gli amartomi rappresentano l'1,2% delle neoplasie benigne. La prevalenza degli amartomi polmonari è stimata in circa lo 0,25% della popolazione generale e rappresenta fino all'8% di tutte le neoplasie polmonari. La maggior parte degli amartomi polmonari viene diagnosticata incidentalmente in pazienti di età compresa tra 40 e 70 anni, ma sono molto rari nella pratica pediatrica.
In generale, la maggior parte degli amartomi viene diagnosticata negli uomini, anche se a livello renale sono più comuni nelle donne e vengono identificati in età adulta.
Circa il 5% dei tumori benigni al seno sono amartomi e colpiscono più comunemente le donne di età superiore ai 35 anni.
L'80-90% delle lesioni amartomatose del cervello e oltre il 50% degli amartomi del cuore sono associati alla sclerosi tuberosa.
Le cause amartomi
I gamartomi appartengono alle malformazioni congenite e sono formazioni di carattere benigno, che si formano da tessuti mesenchimali originati da foglietti germinativi. Le cause della loro insorgenza sono associate alla divisione cellulare incontrollata di tessuti citologicamente normali (connettivo, muscolo liscio, grasso o cartilagine), caratteristici di una determinata sede anatomica, e alla loro crescita eccessiva focale durante l'embriogenesi di quasi tutti gli organi o strutture anatomiche.
La comparsa di più amartomi nello stesso paziente è spesso definita amartomatosi o amartoma pleiotropico.
Questi tumori possono presentarsi sporadicamente o in presenza di alcune malattie ereditarie autosomiche dominanti, nonché di sindromi geneticamente determinate.
In molti casi, gli amartomi si formano quando una rara malattia genetica di natura multisistemica - la sclerosi tuberosa - si manifesta poco dopo la nascita, o nella malattia familiare di Recklinghausen - la neurofibromatosi di tipo 1. [ 2 ]
Fattori di rischio
I principali fattori di rischio per la formazione di amartomi includono la presenza nella storia dei pazienti delle cosiddette sindromi genetiche della poliposi amartomatosa, tra cui:
- Sindrome degli amartomi multipli - sindrome di Cowden, in cui si formano amartomi multipli di origine ecto-, ento- e mesodermica, si osservano poliposi gastrointestinale e manifestazioni mucocutanee;
- Sindrome di Peutz-Jeghers-Turen (caratterizzata dallo sviluppo di polipi amartomatosi benigni nel tratto gastrointestinale);
- Sindrome di Proteo;
- Sindrome di Weil - poliposi giovanile del colon;
- Sindrome di Bannayan-Riley-Ruvalcaba, che, come la sindrome di Cowden, provoca molteplici amartomi (polipi amartomatosi) dell'intestino;
- Sindrome di Carney-Stratakis e complesso di Carney.
Inoltre, gli amartomi si formano nei pazienti con sindrome di Watson ereditaria e nei casi di sindrome di Pallister-Hall sporadica o congenita con amartoma ipotalamico e polidattilia.
Patogenesi
Il meccanismo di aumentata proliferazione dei tessuti germinali con formazione di malformazioni simil-tumorali in vari organi è spiegato da aberrazioni cromosomiche e mutazioni genetiche che possono verificarsi spontaneamente o essere ereditarie.
Nella sclerosi tuberosa, sono state identificate mutazioni nei geni TSC1 o TSC2, oncosoppressori che prevengono e inibiscono la proliferazione eccessiva, ovvero una crescita e una divisione cellulare troppo rapide o incontrollate. E nella neurofibromatosi di tipo 1 e nella sindrome di Watson, mutazioni germinali del gene oncosoppressore mitocondriale NF1.
Nella sindrome tumorale amartomatica, che combina le sindromi di Cowden, Protea, Bannayan-Riley-Ruvalcaba e poliposi giovanile, la patogenesi è associata alla mutazione del gene PTEN, che codifica un enzima coinvolto nella regolazione della proliferazione ed è considerato un gene soppressore del tumore.
Mutazioni nel gene STK11, che codifica per la struttura e la funzione di uno degli enzimi transmembrana della serina, che ne riduce la capacità di inibire la divisione cellulare, portano alla sindrome di Peutz-Jeghers-Turen, con lo sviluppo di polipi intestinali e lesioni cutanee pigmentate. Una mutazione nel gene GLI3, un fattore di trascrizione coinvolto nella formazione del tessuto intrauterino, è stata identificata nella sindrome di Pallister-Hall.
Pertanto, la crescita incontrollata delle cellule dovuta a mutazioni genetiche porta alla formazione di amartomi.
Sintomi amartomi
A seconda della localizzazione degli amartomi si distinguono i loro tipi, ognuno dei quali ha una propria struttura e sintomatologia.
Amartoma del polmone
L'amartoma polmonare può formarsi in qualsiasi lobo e nelle parti periferiche dei polmoni ed è costituito da tessuti normali presenti nei polmoni: adiposo, epiteliale, fibroso e cartilagineo. Nell'80% dei casi, predomina la componente condroide (cellule della cartilagine ialina) con inclusione di adipociti - cellule del tessuto adiposo e cellule epiteliali delle vie aeree. [ 3 ]
I nomi precedenti: amartoma condroide, mesenchimoma, amartoma condromatoso o amartocondroma non sono attualmente raccomandati dall'OMS.
L'amartoma cistico mesenchimale del polmone, d'altro canto, è meno comune e nella maggior parte dei pazienti è associato alla sindrome di Cowden.
La lesione amartomatosa del polmone può non manifestarsi, ma può causare sintomi sotto forma di tosse cronica (spesso con emottisi), respiro sibilante durante la respirazione e difficoltà respiratorie. [ 4 ]
Un amartoma del cuore
I tumori cardiaci primari benigni negli adulti includono l'amartoma dei miociti maturi e, nei neonati e nei bambini con sclerosi tuberosa, il rabdomioma, cioè l'amartoma miocardico dei ventricoli o del setto interventricolare. [ 5 ]
L'amartoma cardiomiocitario maturo si sviluppa nella parete ventricolare (e raramente negli atri) e può apparire come lesioni multiple, masse dense strettamente associate al miocardio sottostante. Il tumore può causare sintomi di insufficienza cardiaca: dolore toracico, palpitazioni e aritmie, soffi cardiaci, edema, dispnea, cianosi.
I rabdomiomi cardiaci, la maggior parte dei quali viene diagnosticata entro il primo anno di vita, sono composti da tessuto muscolare cardiaco formato da mioblasti embrionali e hanno l'aspetto di masse focali solide senza capsula.
In genere, questi amartomi si manifestano in modo asintomatico e regrediscono spontaneamente prima dei 4 anni di età.
Le lesioni amartomatose sono anche considerate da alcuni esperti associate al mixoma complesso di Carney del cuore. [ 6 ]
Gamartoma del tratto gastrointestinale
L'amartoma gastrico è una massa mesenchimale sotto forma di polipo iperplastico epiteliale dello stomaco, polipo di Peutz-Jeghers e raro amartoma mioepiteliale con fasci muscolari lisci ipertrofici. Altri nomi per questo amartoma includono amartoma mioghiandolare, amartoma adenomiomatoso e adenomioma gastrico. Le manifestazioni cliniche tipiche includono dispepsia, dolore epigastrico e sanguinamento del tratto gastrointestinale superiore. [ 7 ], [ 8 ]
Maggiori informazioni nel materiale - Poliposi gastrica
Un amartoma intestinale è un polipo amartomatoso o iperplastico dell'intestino crasso, diagnosticato come adenoma adenomatoso o tubulare. Quando l'amartoma è localizzato nella ghiandola di Brunner del duodeno, i sintomi si manifestano con dolore nella regione epigastrica; nausea, vomito e flatulenza (indicando ostruzione intestinale); e, se di dimensioni considerevoli, sanguinamento gastrointestinale. In caso di amartoma mioepiteliale dell'ileo, i pazienti lamentano dolore addominale, hanno una perdita di peso corporeo e sviluppano anemia cronica. [ 9 ], [ 10 ]
Leggi anche - Polipi rettali
L'amartoma retrorettale è un amartoma cistico o cisti multicamerale dello spazio retrorettale (il tessuto connettivo lasso tra il retto e la sua fascia) che si verifica più comunemente nelle donne di mezza età. Ha l'aspetto di una cisti che fuoriesce dalla parete posteriore del retto, che è rivestita da epitelio e contiene fibre muscolari lisce disposte in modo caotico. Questo amartoma si presenta con dolore addominale inferiore e stitichezza ricorrente. [ 11 ], [ 12 ]
Amartomi del fegato e della milza
L'amartoma biliare multiplo del fegato è un amartoma dei dotti biliari intraepatici interdollici associato a malformazioni del loro sviluppo durante il periodo embrionale. Questo amartoma (singolo o multiplo) è costituito da gruppi dilatati casualmente di dotti biliari e stroma fibrocollageno. [ 13 ]
Gli amartomi biliari sono asintomatici e vengono solitamente scoperti incidentalmente (durante l'esame radiologico o laparotomia). [ 14 ]
Una neoplasia primitiva rara e spesso rilevata incidentalmente di carattere benigno è l'amartoma della milza, costituito da elementi della polpa rossa della milza, sotto forma di una massa omogenea ben definita di consistenza soda. Questa malformazione può essere singola o multipla; comprimendo il parenchima splenico, si può avvertire una sensazione di fastidio e dolore nella zona sottocostale sinistra. [ 15 ], [ 16 ]
Amartomi renali
L'amartoma renale più comune viene diagnosticato come angiomiolipoma renale, poiché questo tumore benigno è costituito da tessuto adiposo maturo con fibre muscolari lisce e vasi sanguigni incorporati. Si forma nella sclerosi tuberosa nel 40-80% dei casi. L'aumento delle dimensioni dell'amartoma (oltre 4-5 cm) porta a dolore e alla comparsa di sangue nelle urine. [ 17 ], [ 18 ]
Amartoma del seno
Le definizioni diagnostiche di amartoma mammario accettate dall'OMS sono termini come adenolipoma, condrolipoma e amartoma mioide. Sebbene spesso chiamato fibroadenolipoma dai mammologi, poiché la formazione tumorale contiene cellule di tessuto fibroso, ghiandolare e adiposo racchiuse in una sottile capsula di tessuto connettivo con contorni distinti. Alla visualizzazione possono essere osservate calcificazioni focali. In questo caso, le manifestazioni cliniche sono assenti. [ 19 ], [ 20 ]
Leggi anche - Tumori al seno
Amartomi del cervello
Un terzo dei pazienti con sclerosi tuberosa presenta un amartoma cerebrale sotto forma di escrescenze corticali intracraniche o tubercoli in vari lobi - al confine tra sostanza grigia e sostanza bianca - o noduli subependimali lungo le pareti dei ventricoli cerebrali. Può anche formarsi un amartoma astrocitico, un astrocitoma subependimale a cellule giganti con rottura corticale, neuroni dismorfici e grandi cellule gliali del parenchima cerebrale (astrociti). I sintomi degli amartomi cerebrali includono attacchi convulsivi e ritardo mentale nei bambini. [ 21 ], [ 22 ]
Una rara malformazione che si verifica durante l'embriogenesi ed è presente alla nascita è l'amartoma ipotalamico, una massa di neuroni eterotopici e cellule gliali. Man mano che il cervello del bambino cresce, il tumore si ingrandisce ma non si diffonde ad altre regioni cerebrali. [ 23 ], [ 24 ]
Se si formano tessuti ipertrofici nella parte anteriore dell'ipotalamo (tuber cinereum), dove è attaccata l'ipofisi, la malformazione manifesta sintomi di sviluppo sessuale prematuro centrale (prima degli 8-9 anni di età): comparsa di eruzioni cutanee acneiche, sviluppo precoce delle ghiandole mammarie e menarca precoce nelle bambine; peli pubici precoci e mutazione della voce nei bambini.
Quando gli amartomi si formano nella parte posteriore dell'ipotalamo, possono verificarsi anomalie nell'attività elettrica del cervello, che nella prima infanzia si manifestano con convulsioni e in una fase successiva (dai 4 ai 7 anni) con epilessia con crisi epilettiche focali con risate improvvise o con pianto involontario, crisi atoniche e tonico-cloniche, nonché crisi di aggressività, problemi di memoria e cognitivi.
L'amartoma ipofisario è un adenoma ipofisario benigno che si verifica sporadicamente.
Gli adulti di mezza età affetti dalla sindrome di Cowden possono presentare una rara massa simil-tumorale, un amartoma del cervelletto, diagnosticato come gangliocitoma cerebellare displastico o malattia di Lhermitte-Duclos. I sintomi possono essere assenti o manifestarsi come cefalea, vertigini, difficoltà di coordinazione dei movimenti e paralisi di singoli nervi cranici.
Amartoma del linfonodo
Quando le cellule del muscolo liscio e del tessuto adiposo, così come i vasi sanguigni e lo stroma collagenoso dei linfonodi inguinali, retroperitoneali, sottomandibolari e cervicali crescono eccessivamente, si forma un amartoma angiomiomatoso di un linfonodo o amartoma angiomiomatoso nodulare - con sostituzione parziale o completa del suo parenchima. [ 25 ], [ 26 ]
Un amartoma della pelle
In presenza di sclerosi tuberosa o neurofibromatosi si osservano vari amartomi della pelle, più spesso sotto forma di macchie ipopigmentate; macchie color caffè e latte; angiofibroma (su guance, mento, pieghe naso-labiali); macchie zigrinate di varia localizzazione (che sono nevi del tessuto connettivo); placche fibrose sulla fronte, sul cuoio capelluto o sul collo.
Una rara manifestazione dermatologica della sclerosi tuberosa (soprattutto negli uomini) è l'amartoma follicolocistico e del collagene, caratterizzato da abbondante deposizione di collagene nel derma, fibrosi perifollicolare concentrica e cisti sottocutanee a forma di imbuto piene di cheratina osservate all'esame istopatologico. [ 27 ]
Con amartomi costituiti da melanociti (cellule che producono il pigmento melanina) la maggior parte degli esperti fa riferimento anche a varie neoplasie melanocitarie, in particolare ai nevi melanocitari congeniti, che rappresentano un'anomalia dell'embriogenesi.
Dal punto di vista eziologico, gli amartomi composti da tessuto vascolare sono anche emangiomi della pelle.
I pazienti con sindrome di Peutz-Jeghers-Thuren presentano un amartoma sotto forma di pigmentazione a chiazze della pelle e delle mucose - lentiginosi periorificiale
I casi di amartoma ectodermico-mesodermico papulare lineare (Hamartoma moniliformis) mostrano un'eruzione cutanea papulare lineare color carne sulla testa, sul collo e sulla parte superiore del torace.
L'amartoma sebocitico è un amartoma delle ghiandole sebacee, per saperne di più leggi la pubblicazione - nevo sebaceo.
Amartoma dell'occhio
Le lesioni amartomatose pigmentate dell'iride nella neurofibromatosi di tipo 1 e nella sindrome di Watson, sotto forma di ammassi nodulari di melanociti dendritici, sono definite amartomi dell'iride o noduli di Lisch. Si tratta di papule trasparenti (di solito non compromettono la vista), arrotondate e a forma di cupola, di colore giallo-marrone, che sporgono dalla superficie dell'iride.
I pazienti con angiofibroma giovanile del rinofaringe e poliposi adenomatosa familiare sviluppano spesso un amartoma combinato della retina e dell'epitelio pigmentato della retina, sotto forma di una macchia nera sulla parte centrale (maculare) della retina. [ 28 ]
Un amartoma del naso
L'amartoma nasale è definito dagli specialisti come amartoma condrosenchimale nasale o condroma nasale, dovuto alla proliferazione benigna dell'epitelio respiratorio, delle ghiandole sottomucose e del mesenchima condro-osseo. Le sue manifestazioni cliniche dipendono dalle dimensioni e dalla localizzazione della lesione e includono: congestione nasale, difficoltà nella respirazione nasale e nell'allattamento al seno nei neonati, secrezione nasale chiara e acquosa e sanguinamento nasale. Un amartoma può crescere con il bambino e diffondersi nelle orbite oculari, causando lo spostamento in avanti o indietro del bulbo oculare, strabismo o disturbi oculomotori. [ 29 ]
Un amartoma in un bambino
Tutte le lesioni amartomatose sopra menzionate di vari organi e strutture anatomiche sono presenti nei bambini con sindromi corrispondenti.
I neonati presentano un amartoma mesenchimale della parete toracica o un amartoma cartilagineo della costa, masse solide immobili derivanti dalla crescita focale eccessiva di elementi scheletrici normali con elementi cartilaginei, vascolari e mesenchimali. Questo amartoma può causare insufficienza respiratoria e lo sviluppo di sindrome da distress respiratorio. L'amartoma mesenchimale del fegato è il secondo tumore epatico benigno più frequente nei bambini. Questa formazione simil-tumorale (più spesso localizzata nel lobo destro dell'organo) è costituita da cellule dello stroma mesenchimale, epatociti e cellule epiteliali del rivestimento dei dotti biliari. Il quadro clinico include una massa palpabile nella cavità addominale, anoressia e perdita di peso e, in caso di dimensioni significative (fino a 10 cm e oltre), il tumore ricopre i dotti biliari extraepatici e la vena cava inferiore, causando ittero ed edema degli arti inferiori.
Un amartoma è un nefroma mesoblastico congenito (che si verifica in 1 neonato su 200.000) che può causare gonfiore addominale nel neonato con una massa palpabile di consistenza densa nel quadrante superiore destro dell'addome. I neonati possono anche presentare respiro rapido e superficiale.
Tra le anomalie congenite rare rientra l'amartoma fibroso dell'infanzia, che si manifesta nei bambini nei primi due anni di vita e si presenta come una massa nodulare indolore nei tessuti sottocutanei di ascella, collo, spalla e avambraccio, schiena e torace, coscia, piede e genitali esterni.
L'amartoma angiomatoso eccrino in un bambino può essere presente alla nascita o manifestarsi nella prima infanzia. Questo tumore benigno di natura amartomatosa si presenta solitamente sotto forma di noduli e/o placche bluastre o brunastre, derivanti dalla proliferazione del tessuto delle ghiandole sudoripare eccrine e dei capillari negli strati medi e profondi del derma. Questo amartoma può causare iperidrosi localizzata e aumento della crescita dei peli.
Complicazioni e conseguenze
È generalmente riconosciuto che gli amartomi raramente recidivano o si trasformavano in tumori maligni. Spesso mostrano sintomi lievi o assenti e talvolta scompaiono nel tempo. Ma nei casi più gravi e a seconda della sede di formazione, queste malformazioni possono avere gravi complicazioni e conseguenze.
Innanzitutto, un amartoma può crescere fino a raggiungere dimensioni tali da iniziare a premere sui tessuti e sugli organi circostanti, compromettendone le funzioni.
L'amartoma cardiaco nei bambini può causare anomalie persistenti del ritmo cardiaco, difetti valvolari e alterazione del flusso sanguigno intracardiaco con conseguente insufficienza cardiaca congestizia.
Le complicazioni dei polipi amartomatosi del tratto gastrointestinale sono emorragia gastrointestinale, ostruzione e intussuscezione intestinale (con possibile esito fatale). Un amartoma renale di grandi dimensioni può provocare la rottura del rene.
Un amartoma nel cervello può causare la sindrome da idrocefalo ostruttivo.
Negli amartomi ipotalamici e ipofisari, la produzione di ormone somatotropo (ormone della crescita) può essere compromessa, portando allo sviluppo di nanismo ipofisario (ipopituitarismo) nei bambini. Gli amartomi ipotalamici nei bambini possono anche portare a epilessia farmacoresistente.
Le complicazioni dell'amartoma dell'epitelio pigmentato della retina sono rappresentate da disfunzione della retina e/o del nervo ottico, edema maculare, neovascolarizzazione della coroide e distacco della retina.
Diagnostica amartomi
Una parte importante della diagnosi degli amartomi e delle sindromi correlate è la raccolta dell'anamnesi, inclusa la storia familiare.
Gli esami di laboratorio includono esami del sangue: esami clinici generali; elettroliti sierici; profilo linfocitario; livelli di calcio, potassio, fosfato e urea; e test di funzionalità epatica. Se possibile, viene eseguita una biopsia con agoaspirato sottile della massa, poiché l'esame istologico è fondamentale per la diagnosi e la scelta del trattamento.
La diagnostica strumentale consente la visualizzazione della formazione tumorale amartomatosa e l'identificazione della sua esatta localizzazione, per la quale vengono utilizzati raggi X, angiografia, elettroencefalografia (EEG), ultrasuoni (ecografia), TC (tomografia computerizzata), PET (tomografia a emissione di positroni), RM (risonanza magnetica).
Diagnosi differenziale
In caso di masse anomale, la diagnosi differenziale è molto importante. Pertanto, si differenziano tubercoloma e amartoma; amartoma polmonare e carcinoma polmonare primario, carcinoide broncogeno, malattia metastatica. L'amartoma cerebrale deve essere distinto dal craniofaringioma e dal glioma ipotalamico-chiasmatico. La diagnosi differenziale dell'amartoma come nefroma mesoblastico congenito include il tumore di Wilms (nefroblastoma maligno), il sarcoma a cellule chiare del rene e il tumore renale ossificante nei neonati.
Chi contattare?
Trattamento amartomi
Se l'amartoma è asintomatico e viene scoperto accidentalmente, non è necessario alcun trattamento, ma è necessario monitorarne il "comportamento" e le condizioni del paziente. In altri casi, la terapia mira a ridurre l'intensità dei sintomi e a prevenire le complicanze. Ad esempio, nell'amartoma ipotalamico con sintomi di pubertà precoce, vengono prescritti alcuni farmaci che inibiscono il rilascio di determinati ormoni. I farmaci cardiaci vengono utilizzati per trattare i sintomi dell'insufficienza cardiaca nei pazienti con amartomi cardiaci.
L'asportazione chirurgica degli amartomi è indicata per confermare la diagnosi e nei casi di sintomi intensi non correggibili clinicamente.
Ad esempio, gli amartomi polmonari possono essere asportati mediante resezione a cuneo e, nei casi più gravi, mediante l'asportazione di un lobo polmonare (lobectomia). Anche un amartoma mammario può essere asportato e, se di grandi dimensioni, potrebbe essere necessaria una mastectomia parziale o completa.
La termoablazione stereotassica a radiofrequenza o l'ablazione laser possono essere utilizzate per rimuovere i polipi amartomatosi. Viene utilizzata anche la radiochirurgia con raggi gamma altamente focalizzati (Gamma Knife) per gli amartomi ipotalamici o amartomi astrocitari.
Prevenzione
L'unico metodo per prevenire lo sviluppo di amartomi può essere considerato lo screening genetico dei futuri genitori del bambino.
Previsione
La prognosi complessiva di questa anomalia congenita dipende dalla localizzazione e dalle dimensioni della neoplasia, nonché dalle comorbilità e dallo stato di salute generale del paziente.